Skeletal Dysplasias
INTRODUCTION
Skeletal dysplasias are heritable disorders of bone caused by abnormal development, growth, and maintenance of the human skeleton. They are a heterogeneous group of conditions that vary significantly in clinical severity, ranging from conditions that almost always cause death in utero or soon after to birth, to conditions resulting in short stature and chronic health complications that are not generally life limiting. This chapter presents an approach to the diagnosis and management of the skeletal dysplasias that commonly present in the neonatal period.
EPIDEMIOLOGY
The overall birth prevalence of all types of skeletal dysplasias is estimated to be 2–3 per 10,000 births.1,2 The 2010 revision of the Nosology and Classification of Genetic Skeletal Disorders recognized 456 different conditions and classified them into different groups by their clinical and radiographic features and molecular pathogenesis.3 Skeletal dysplasias classified as lethal (those that result in death in utero or early neonatal death) make up about 50%. The most common groups of disorders are osteogenesis imperfecta (various types), disorders related to fibroblast growth factor receptor 3 (FGFR3) disorders (thanatophoric dysplasia and achondroplasia are most common), and type II collagenopathies (achondrogenesis is most common) (see further sections for descriptions of these disorders).1 Achondroplasia is the most common cause of disproportionate short stature that is not associated with in utero or early neonatal death. The prevalence of achondroplasia has been estimated at 0.36–0.6 per 10,000 live births (1/27,780–1/16,670 live births).4
PATHOGENESIS
Identification of the genes responsible for many of the skeletal dysplasias has led to better understanding of the underlying pathogenesis and emerging treatments of these disorders.
Defects in Local Regulation of Cartilage Growth
Defects in local regulation of cartilage growth include disorders caused by abnormalities in growth factors and their receptors. FGFR3 plays a role in the negative regulation of bone growth by inhibiting cell growth in cartilaginous growth plates. Almost all cases of achondroplasia are caused by 1 of 2 specific gain-of-function mutations in the FGFR3 gene, which result in upregulation of the FGFR3 pathway. The lethal thanatophoric dysplasias (type I and II) are caused by different mutations in the FGFR3 gene.
Defects in Structural Proteins of Cartilage
Defects of structural cartilage proteins such as collagen type I, II, IX, X, and XI and extracellular matrix proteins such as COMP (cartilage oligometric matrix protein) result in various different forms of skeletal dysplasias. Mutations in the gene encoding collagen type II cause the group of disorders known collectively as the type II collagenopathies, which comprise achondrogenesis type II, hypochondrogenesis, spondyloepiphyseal dysplasia congenita (SEDC), Kneist dysplasia, and Stickler syndrome.
Defects in Cartilage Metabolic Pathways
Defects of enzymes, ion channels, and transporters essential for cartilage metabolism and homeostasis have been identified as the cause of other skeletal dysplasias.
TRPV4
The transient receptor potential cation channel, subfamily V, member 4 (TRPV4) gene encodes for a calcium-permeable ion channel, transient receptor potential cation channel subfamily V, member 4. Dominant mutations in this gene result in activation of the channel, leading to increased concentration of calcium within chondrocytes. Mutations in TRPV4 are the cause of a number of skeletal dysplasias, including lethal and nonlethal metatropic dysplasia, which present prenatally or in the neonatal period and spondylometaphyseal dysplasia Kozlowski type, which presents in childhood.5 The variable severity of clinical features seen in the TRPV4 groups of skeletal dysplasias can be correlated with the degree of activation of the channel.6 Interestingly, other mutations in the TRPV4 gene can present with an arthritic phenotype predominantly involving the hands and feet.7
Diastrophic Dysplastic Sulfate Transporter
The diastrophic dysplastic sulfate transporter (DTDST) gene encodes for a sulfate transporter (SLC26A2). Recessive mutations in this gene result in impairment of sulfate transport into the chondrocytes. DTDST mutations are the cause of the lethal disorders achondrogenesis IB and atelosteogenesis II, the nonlethal but severe disorder diastrophic dysplasia, and the much milder condition autosomal recessive multiple epiphyseal dysplasia. The variation in the severity of clinical features is correlated with the degree of impairment of transport of sulfate. The more severe phenotypes are caused by null mutations in both alleles of the gene, whereas the milder phenotypes are caused by mutations that only partially impair sulfate transport.8
DIFFERENTIAL DIAGNOSIS
Making a diagnosis of a specific skeletal dysplasia is often difficult given the vast number of recognized types. The involvement of physicians with specific skills in skeletal dysplasias, such as clinical geneticists, orthopedic specialists, and pediatric radiologists, is essential when trying to make an accurate diagnosis in the neonatal period.
Based on clinical history and examination, family history, and expert evaluation of radiology (the single most powerful diagnostic tool), it is usually possible to make a diagnosis in the majority of those who present in the neonatal period with a skeletal dysplasia. Molecular testing can provide confirmation of a clinical diagnosis. In some cases, a skeletal dysplasia will be unclassifiable in the neonatal period, but with time, the development of additional clinical or radiographic features will allow a specific diagnosis to be made.
Including all possible diagnoses is beyond the scope of this chapter, so examples of the more commonly occurring conditions are given to illustrate the different presentations in the neonatal period. Key radiographic findings are discussed, but given the complexity of the skeletal changes for each type of dysplasia, it is not possible to provide extensive details of the radiographic findings for each condition
“Lethal” Skeletal Dysplasias
“Lethal” skeletal dysplasias are those that lead to death in utero or shortly after birth because of respiratory insufficiency. They will generally have been detected antenatally because of characteristic ultrasound findings. These conditions are presented first in each section.
Infant With Short Limbs
Thanatophoric Dysplasia (Types 1 and 2)
Thanatophoric dysplasias are characterized by very short limbs, a relatively normal trunk length, and narrow thorax. The head is relatively large, and craniosynostosis (cloverleaf skull deformity) occurs more commonly in type 2. Radiographs show short ribs; narrow thorax; severe flattening of the ossification centers of the vertebral bodies (platyspondyly); short, broad pelvic bones; and short, broad femora, which are bowed in type 1 and straight in type 2. Both types of thanatophoric dysplasia are due to dominant mutations in the fibroblast growth factor receptor 3 (FGFR3) gene. Long-term survival may be possible with invasive respiratory support but is associated with a poor long-term outcome with severe cognitive impairment (often secondary to temporal lobe dysplasia), ventilator dependence, and a final height of 80–90 cm.
Fibrochondrogenesis
Fibrochondrogenesis is an autosomal recessive disorder caused by mutations in the type XI collagen gene. It is characterized by midfacial hypoplasia, short nose with anteverted nares, micrognathia, short limbs, and small thorax. Radiographs show platyspondyly (flattened vertebrae) with ossification defects of the posterior aspects of the vertebrae, short long tubular bones with bulbous metaphyseal ends, and short ribs with cupped ends.
Atelosteogenesis (Types I, II, III)
Type I: Infants have rhizomelic limb shortening, midfacial hypoplasia, short broad hands, and talipes equinovarus. Radiographic changes include distal hypoplasia of humerus and femur; short bowed radius/ulna and fibular hypoplasia; broad short tubular bones; absent ossification of metacarpals and distal/middle phalanges; and hypoplastic vertebrae with coronal clefts of the bodies.
Type III: The facial features in type III are similar to those seen in type I. Multiple joint dislocations of elbows, hips, and knees are present. Hand and feet changes include broad distal phalanges, syndactyly, camptodactyly of fingers and toes, and talipes equinovarus.
Type I and III atelosteogenesis are allelic disorders caused by dominant mutations in filamin B. Long-term survival is possible in type III.
Type II: Clinical features include relative macrocephaly, short trunk, small chest, short limbs with hitchhiker thumbs, sandal gap between first and second toe, and talipes equinovarus. Typical facial features include midfacial hypoplasia, micrognathia, and cleft palate. Type II atelosteogenesis is caused by mutations in the diastrophic dysplasia sulfate transporter (DTSDT) gene.
Nonlethal Skeletal Dysplasias
Achondroplasia
Achondroplasia is the most common nonlethal skeletal dysplasia causing disproportionate short stature. The clinical features are well recognized and allow a diagnosis to be made in most infants in the neonatal period. Infants have short limbs with rhizomelic segments relatively shorter than other limb segments (which are also short), relative macrocephaly, kyphosis at the thoracolumbar junctions, “trident” hand configuration, and characteristic facial features that include depressed nasal bridge, mild midfacial hypoplasia, and frontal bossing. It is common to see loose redundant skin folds in the limbs, which reflect normal soft tissue growth over shortened bones. In infancy, the signs on radiographs, which allow confirmation of a clinical diagnosis, include characteristic changes in the pelvis (squared iliac wings, narrow sacrosciatic notches, flat acetabular margins); reduction in the interpediculate distance from the upper to lower lumbar vertebrae (distance normally widens); and a typical area of radiolucency at the proximal femur (Figure 62-1A–1C). Achondroplasia is caused by mutations in the FGFR3 gene, and in the majority of cases (80%), these are de novo.
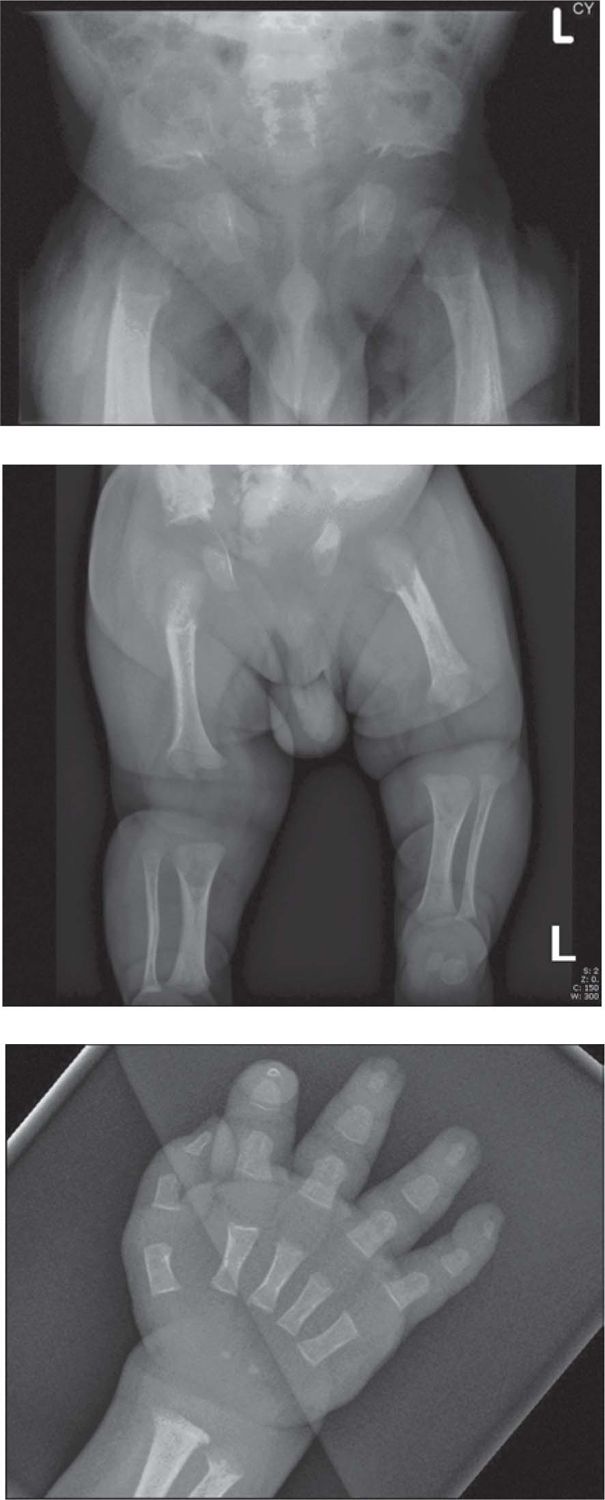
FIGURE 62-1 A, Pelvis radiograph of an infant with achondroplasia. Note reduction in the interpediculate distance from the upper to lower lumbar vertebrae, squared iliac wings, and areas of radiolucency at the proximal femora. B, Lower-limb radiograph of an infant with achondroplasia. Note shortening of the long bones with the rhizomelic segments relatively shorter than the other segments and redundant skin folds seen in the soft tissues. C, Radiograph of the hand of an infant with achondroplasia. Note short, broad proximal bullet-shaped middle and proximal phalanges and separation of the fingers, giving the “trident” appearance to the hand.
Ellis van Creveld Syndrome (Chondroectodermal Dysplasia)
Ellis van Creveld syndrome (chondroectodermal dysplasia) causes short limbs with progressive shortening of proximal-to-distal segments and short ribs, leading to narrow thorax. Polydactlyly of fingers and often toes is a feature. Ectodermal abnormalities, including hypoplastic nails and dental anomalies (eg, neonatal teeth are also seen). The upper lip is short and connected to the alveolar ridge by multiple frenulae. Structural cardiac abnormalities are present in 50% of infants. This syndrome is an autosomal recessive condition caused by mutations in the EVC gene. Parents should be examined as they can display minor dental, hand, and feet anomalies (Weyers acrofacial dysostosis).
Metatropic Dysplasia
Infants with metatropic dysplasia usually have normal length because of their long trunk with short limbs and a narrow chest. A tail-like caudal appendage overlying the sacrum is a common feature. The joints are prominent and often restricted in movement. Radiographs show defective ossification and abnormally shaped vertebral bodies and narrow thorax. A rapidly progressive kyphoscoliosis usually develops in childhood. A lethal form with more severe clinical and radiographic signs also exists. Metatropic dysplasia is caused by dominant mutations in the TRPV4 gene.5
Diastrophic Dysplasia
Clinical features of diastrophic dysplasia include short limbs and short stature at birth, with talipes equinovarus and contractures of other joints present. The thumbs are proximally placed and abducted (“hitchhiker thumb”), and there is an increased gap between the first and second toe. The cystic swellings of the external ears that usually appear within the first 12 weeks of life are pathognomonic for this condition and can ossify. Cleft palate occurs in approximately 50% of cases. In infancy, radiographs show cervical kyphosis; short tubular bones with broad metaphyses; and short, rounded first metacarpals. Diastrophic dysplasia is caused by mutations in the DTDST gene and is inherited as an autosomal recessive trait.
Infants With Bowing/Shortening of Bones
Campomelic Dysplasia
A proportion of infants with campomelic dysplasia will die in the early neonatal period because of respiratory insufficiency secondary to pulmonary hypoplasia (small thorax) and larynotracheomalacia from deficient airway cartilage. Some, but not all, long-term survivors have cognitive impairment. The clinical features include bowing of femora and tibia with pretibial skin dimples; micrognathia; dysmorphic facial features (flat nasal bridge, low-set ears, long philtrum); dislocated hips; and talipes equinovarus. In up to 75% of XY males with campomelic dysplasia, the external genitalia are either ambiguous or normal female. This is associated with abnormalities of the internal genitalia, with a mixture of Müllerian and Wolffian duct structures present. The key radiographic findings include bowing of the femur and tibia; hypoplastic scapula; small, bell-shaped thorax; 11 pairs of ribs; hypoplastic vertebrae; and narrow iliac wings. Campomelic dysplasia is an autosomal dominant condition caused by mutations in the SOX9 gene.
Hypophosphatasia
Two forms of hypophosphatasia are relevant in the neonatal period. The perinatal lethal form is characterized by absence of ossification of the bones of the skull and limbs, leading to the clinical findings of a soft head and short, deformed extremities. Bony spurs (“Bowdler” spurs) of the midshafts of bones may be palpable. The ribs are short and thin, which leads to respiratory insufficiency, and death usually occurs shortly after birth. The infantile form is characterized by symptoms of hypercalcemia (episodic vomiting, failure to thrive, constipation, irritability, and hypotonia). The cranial sutures are widely patent and fontanelles bulging. The long bones are bowed with associated skin dimples. The ends of the long bones and ribs are swollen. The key radiographic findings include delayed and defective ossification of the skull bones, ribs, and long bones. Paradoxically, given the widely open sutures, craniosynostosis can develop, leading to raised intracranial pressure. With supportive treatment, including management of hypercalcemia and respiratory support, infants survive. The laboratory findings for both conditions are identical, with very low/absent alkaline phosphatase, hypercalcemia, elevated plasma pyridoxal 5′ phosphate, and elevated calcium and phosphoethanolamine in the urine. The disorder is caused by deficiency of the tissue nonspecific alkaline phosphatase enzyme because of mutations in the ALPL gene and is inherited in an autosomal recessive pattern.
Osteogenesis Imperfecta
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


