Severe Combined Immunodeficiency (SCID)
BACKGROUND
Severe combined immunodeficiency (SCID) is a fatal primary immune condition characterized by the absence of both humoral and cellular immunity with severe lymphopenia. Patients with SCID have reduced numbers and function of both T and B lymphocytes (and in some cases, natural killer [NK] cells) and hypogammaglobulinemia. If left untreated, babies with SCID most often die within 1 to 2 years of life because of severe, recurrent infections.
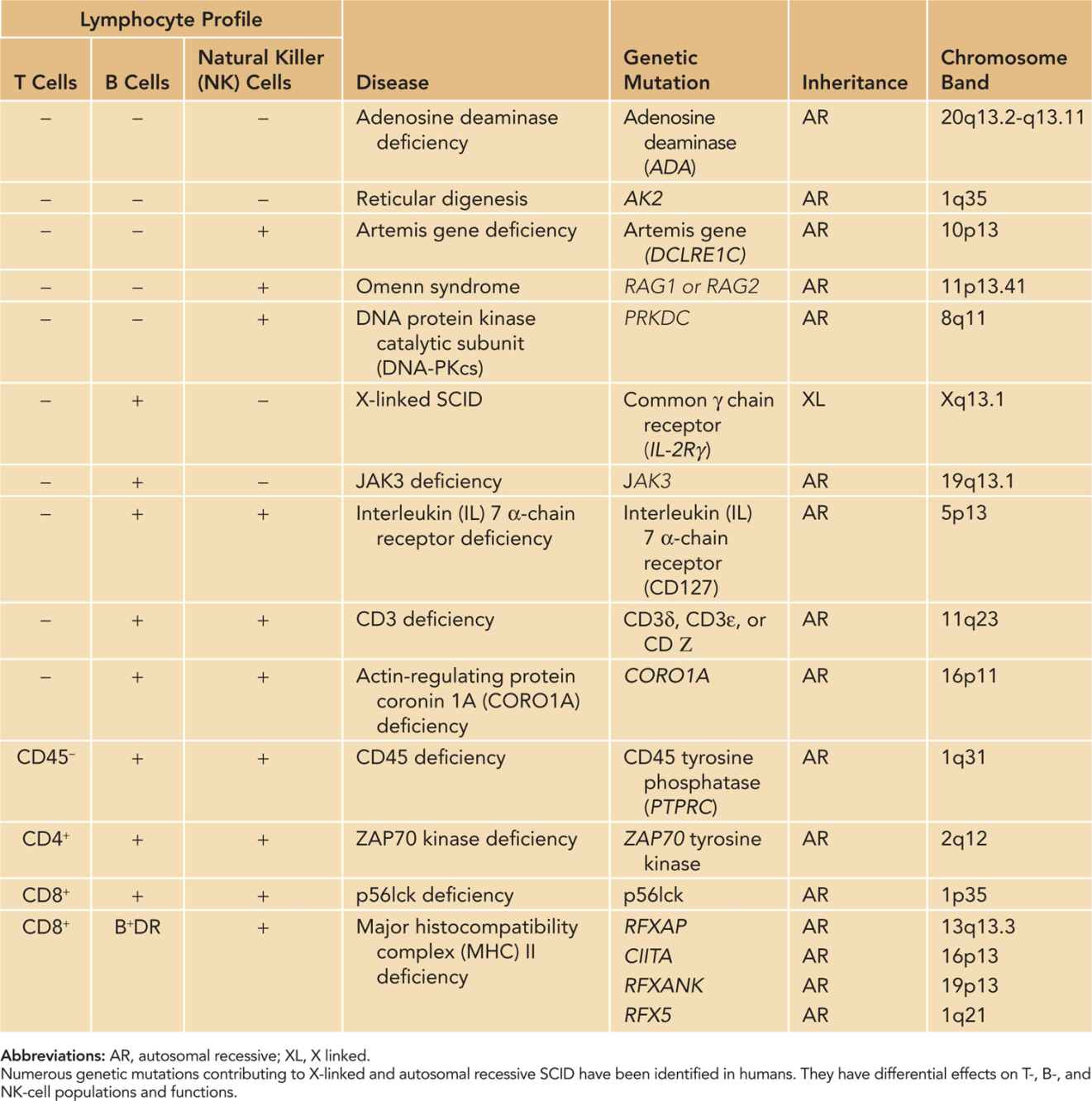
Since SCID was initially described over 50 years ago, 15 specific genetic mutations that contribute to disease development have been identified. The most common type is X-linked SCID (SCID-X1), which accounts for approximately 46% of cases in the United States (Figure 111-1). SCID can also be inherited as an autosomal recessive disease that results from deficiencies in adenosine deaminase (ADA), Janus kinase 3 (JAK3), interleukin 7 receptor α chain (IL7R), recombination activating genes (RAG1 or RAG2), Artemis gene, CD45, and others (Table 111-1).
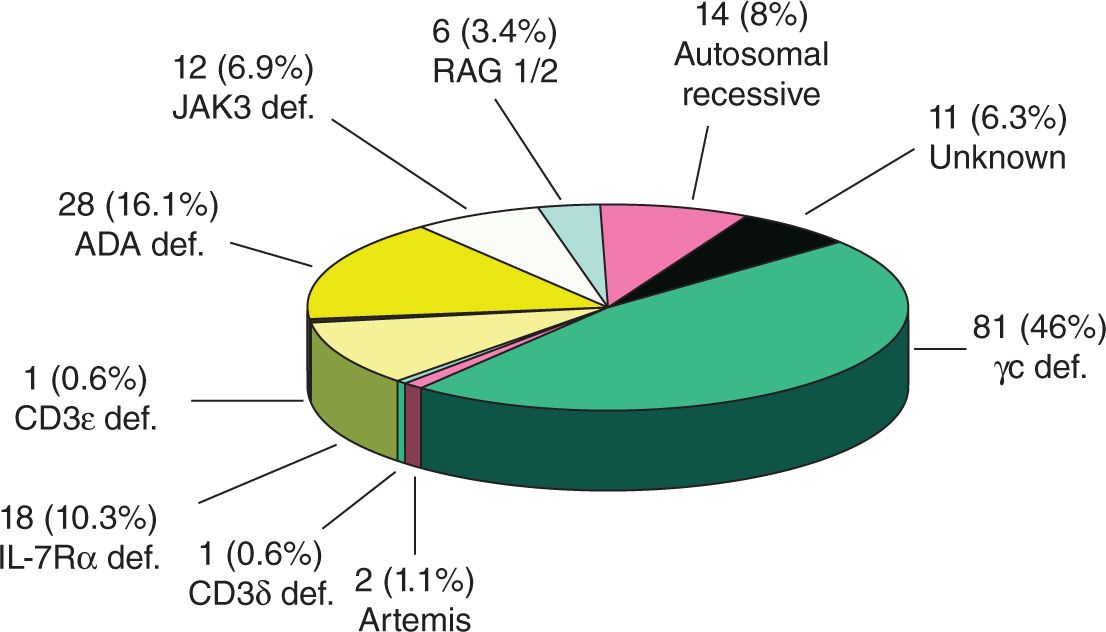
FIGURE 111-1 The relative frequencies of particular genetic mutations were evaluated in 174 consecutive cases of human severe combined immunodeficiency (SCID) evaluated at Duke University over 30 years. The most common type is X-linked SCID, which disables common gamma (γc) signaling. ADA, adenosine deaminase; JAK3, Janus kinase 3; IL7R, interleukin 7 receptor α chain; RAG1 or RAG2, recombination activating genes 1 or 2. (Reproduced with permission from Buckley.1
Table 111-1 Classification of Severe Combined Immunodeficiency (SCID)
PATHOPHYSIOLOGY
X-Linked Recessive SCID
The most prevalent form of SCID, SCID-X1, is characterized by the absence of T and NK lymphocytes and by nonfunctional B lymphocytes. Its defect is mapped to the Xq13 region, and the faulty gene is identified as IL2RG, which encodes the γc chain shared by 6 cytokine receptors that are critical for maintaining immune homeostasis: interleukins (ILs) 2, 4, 7, 9, 15, and 21.
Although SCID-X1 patients lack T- and B-cell function and have low numbers of T or NK cells, they produce normal or elevated numbers of B cells. These B cells are inherently nonfunctional because they fail to produce immunoglobulin even after bone marrow transplantation and restoration of T-cell function. Similarly, NK cells that are present have low-to-absent cell function, and after bone marrow transplantation in patients with SCID-X1, NK-cell generation fails to persist.
Autosomal Recessive SCID Caused by Adenosine Deaminase Deficiency
About half of those affected with autosomal recessive SCID have ADA deficiency, found in about 16% of the total SCID population.1 ADA deficiency was the first identified molecular cause of SCID and involves mutation or deletion of the gene encoding ADA on chromosome 20q13-ter. Because accumulation of ADA substrates or metabolites is toxic to T, B, and NK cells, infants with ADA deficiency have more profound lymphopenia than patients with other types of SCID, with mean absolute lymphocyte counts of less than 500/mm3. A unique symptom to patients with ADA SCID includes multiple skeletal abnormalities of chondro-osseous dysplasia, which occur primarily at the costochondral junctions.
In addition to general SCID treatment and bone marrow transplantation, ADA-deficient patients may undergo enzyme replacement therapy with polyethylene glycol-modified bovine ADA (PEG-ADA). Although this treatment has achieved success in restoring immunocompetence, bone marrow transplantation is still more effective. Therefore, if bone marrow transplantation is being considered, PEG-AGA therapy should not be initiated because it will enable the patient to produce a graft-vs-host response.
Autosomal Recessive SCID Caused by Janus Kinase 3 Deficiency
Mutations in the γ-chain/JAK3 signaling pathway produce symptoms and lymphopenia similar to those in SCID-X1 patients. JAK3 enables T-cell maturation and differentiation, and the common γ chain is also a receptor of IL-15, a key growth factor for NK cells. Consequently, patients with JAK3 deficiency are depleted of T and NK cells. Antibody production by B cells is still severely impaired because of lack of T-cell interaction.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


