Scleroderma
Francesco Zulian
Juvenile scleroderma syndromes are multisystem autoimmune rheumatic diseases whose unifying characteristic is the development of hard skin before age 16. They can be separated into two main categories: those with diffuse skin sclerosis involving many sites of the body together with internal organ involvement (juvenile systemic sclerosis [JSSc]) and those with circumscribed skin induration but no vascular or internal organ involvement (juvenile localized scleroderma [JLS]).
JUVENILE SYSTEMIC SCLEROSIS
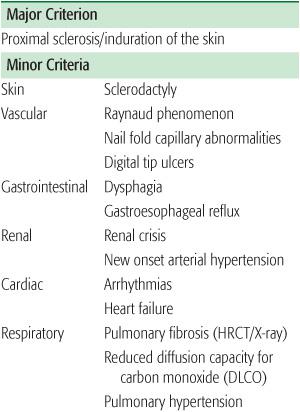
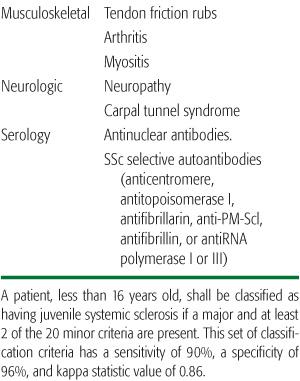
Juvenile systemic sclerosis (JSSc) is a chronic multisystem connective tissue disease characterized by the symmetrical thickening and hardening of the skin, associated with fibrous changes in such internal organs as the esophagus, intestinal tract, heart, lungs and kidneys, plus arthritis and myositis. A Committee on Classification Criteria for JSSc, including pediatricians, rheumatologists, and dermatologists, recently proposed new classification criteria (Table 206-1).1
 EPIDEMIOLOGY
EPIDEMIOLOGY
Systemic sclerosis is a rare condition in any age group, with an estimated annual incidence ranging from 0.45 to 1.9 in 100,000, and a prevalence of approximately 15 to 24 in 100,000.2
Onset in childhood is particularly uncommon: children under age 16 account for less than 5% of all cases,3 and fewer than 10% develop systemic sclerosis before age 20.4,5
Table 206-1. Preliminary Classification Criteria for Juvenile Systemic Sclerosis
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
Children developing scleroderma typically present with Raynaud phenomenon and skin changes (eFig. 206.1  ). Raynaud phenomenon is the first sign of the disease in 70% of patients and in 10% it is complicated by digital infarcts (eFig. 206.2
). Raynaud phenomenon is the first sign of the disease in 70% of patients and in 10% it is complicated by digital infarcts (eFig. 206.2  ). It is more common in the fingers but can be observed in other acral regions including the toes, ears, lips, tongue, and tip of the nose.
). It is more common in the fingers but can be observed in other acral regions including the toes, ears, lips, tongue, and tip of the nose. 
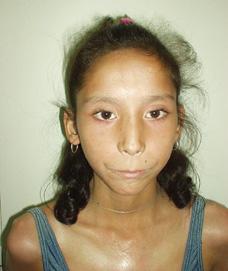
Proximal skin induration usually develops somewhat later and is the second most common complaint, present in 41% of patients at disease onset.6 Cutaneous changes characteristically evolve in a sequence beginning with edema, followed by induration, and eventually resulting in marked skin tightening and joint contractures. The skin becomes waxy in texture, tight, hard, and bound to subcutaneous structures. This is particularly noticeable in skin of the digits and face where the characteristic expressionless appearance of the skin may be the first clue to the diagnosis (Fig. 206-1).
Other presenting complaints include arthralgia, arthritis, and, although less frequently, muscle weakness, dyspnea, and calcinosis.
In children with juvenile systemic sclerosis, visceral organ involvement may be widespread; when it occurs, it is associated with significant morbidity. The gastrointestinal and cardiopulmonary systems are most commonly involved, but effects on the kidneys, peripheral nerves, and musculoskeletal system also can lead to significant discomfort and disability. Cardiorespiratory complications are the leading cause of death in children with juvenile systemic sclerosis.3,7,8
Gastrointestinal involvement occurs in 30% to 70% of children with juvenile systemic sclerosis. Most affected patients have esophageal dysfunction, resulting in gastroesophageal reflux and dysphagia. Manometry, esophageal scintigraphy, and intraesophageal 24-hour pH monitoring provide more sensitive indicators of diminished lower esophageal sphincter tone and gastroesophageal reflux.9 Large bowel involvement is less frequent and presents as alternating complaints of constipation and diarrhea, bloating or abdominal discomfort.
Pulmonary involvement, although frequently asymptomatic, also may present as a dry, hacking cough or as dyspnea on exertion. Other abnormalities associated with juvenile systemic sclerosis may include pleuritis, abnormal diffusion capacity for carbon monoxide (DLCO) (which may be the earliest manifestation of interstitial fibrosis) or pulmonary arterial hypertension.3,6 Unlike adult scleroderma, juvenile systemic sclerosis is infrequently complicated by pulmonary interstitial fibrosis. High-resolution computed tomography (HRCT) may reveal pulmonary disease even in the presence of a normal chest radiograph. In children, HRCT findings include ground glass opacification, subpleural micronodules, linear opacities, and honeycombing.10 Pulmonary hypertension occurs rarely in pediatric scleroderma.
Cardiac involvement is present in around one fifth of pediatric scleroderma patients and represents a primary cause of morbidity among children with juvenile systemic sclerosis.3,10,11 Pericardial effusions are not common and when present are usually of no hemodynamic significance. Pulmonary hypertension caused by pulmonary vascular disease can lead to myocardial damage and right-heart failure.
In a case series of children with systemic sclerosis, about 10% had some kind of renal involvement including either increased urinary protein excretion or an elevated serum creatinine level.6 Although renal involvement in children appears to be less severe or frequent than in adults, the abrupt onset of accelerated hypertension with acute renal failure (scleroderma renal crisis) remains one of the most severe and dangerous complications of juvenile systemic sclerosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


