Rhabdomyosarcoma
Douglas S. Hawkins and Frederic G. Barr
Rhabdomyosarcoma (RMS) is the most common soft tissue tumor in children, representing approximately 5% of all cancers among children and adolescents.1-5 Approximately 350 new cases are diagnosed each year in the United States, for an annual incidence of 4.3 per million children and adolescents younger than 20 years.1 Although RMS can occur in adolescents and adults, two thirds of patients are less than 10 years of age.1,4 The incidence of RMS is slightly higher in males.1 With current multimodal therapy (including chemotherapy, radiotherapy, and surgery), approximately 70% of children with RMS can be cured.2-6
 GENETICS AND EPIDEMIOLOGY
GENETICS AND EPIDEMIOLOGY
A small fraction of RMS cases occur as part of recognizable genetic syndromes, either as an inherited gene in an affected family or as a new germline mutation. These syndromes can be divided into those with only tumor susceptibility and those with both tumor susceptibility and nonneoplastic effects. The former category includes both Li Fraumeni and hereditary retinoblastoma syndromes, which are caused by mutations in the TP53 and RB1 tumor suppressor genes, respectively.7 In Li Fraumeni syndrome, which is characterized by susceptibility to a heterogeneous group of cancers, RMS occurs relatively frequently in addition to other soft tissue sarcomas, breast cancer, brain tumors, and acute leukemia.8 In contrast, RMS occurs less frequently as a secondary tumor in hereditary retinoblastoma syndrome, typically presenting in or near the radiation field following treatment of the primary retinoblastomas.9
Costello syndrome and neurofibromatosis type-1 are syndromes with tumor susceptibility and nonneoplastic effects that are caused by mutations of genes in the RAS signaling pathway—HRAS and NF1, respectively.10 In addition to developmental effects involving the skin and brain, these two syndromes are characterized by a high incidence of benign tumors (skin papillomata and neurofibromas, respectively) and a lower incidence of malignant tumors. RMS is the most common cancer in Costello syndrome, whereas RMS occurs less commonly than malignant peripheral nerve sheath tumors in neurofibromatosis type-1.
In a final group of other tumor susceptibility syndromes with nonneoplastic effects, there are only a few reported RMS cases. These conditions include Beckwith-Wiedemann, Gorlin, and Rubinstein-Taybi syndromes, which are linked to dysregulated imprinted genes in the 11p15 chromosomal region, PTCH mutations, and CREBBP mutations, respectively.7,11,12 In the first two conditions, RMS occurs infrequently relative to a frequent cancer type (Wilms tumor and basal cell carcinoma, respectively), but in the latter, RMS and all other tumors occur infrequently.10,13,14
 PATHOLOGY AND GENETICS
PATHOLOGY AND GENETICS
The tumors of the RMS family have in common a poorly understood relationship to the skeletal muscle lineage and often can be diagnosed based on evidence of this differentiated phenotype. RMS is then further subclassified into two main histopathologic subtypes—alveolar (ARMS) and embryonal (ERMS).15 ARMS is characterized by small cells with round nuclei and scant cytoplasm. Aggregates of ARMS tumor cells are typically divided by fibrovascular septae, and spaces often form within these aggregates to produce an “alveolar” appearance. In contrast, ERMS tumor cells can show varying cellular features that parallel the process of myogenic differentiation, ranging from small round cells to large elongated cells with eccentric oval nuclei and abundant eosinophilic cytoplasm. Furthermore, the overall density of ERMS tumor cells often varies in these tumors with highly cellular areas intermixed with low cellular areas in a loose myxoid stroma.
At the cytogenetic level, most ARMS cases can be distinguished from ERMS as well as other pediatric solid tumors by the presence of recurrent chromosomal translocations.16 The most frequent translocation involves chromosomes 2 and 13 [t(2;13)(q35;q14)], and a less frequent variant involves chromosomes 1 and 13 [t(1;13)(p36;q14)]. These translocations juxtapose portions of the PAX3 or PAX7 gene on chromosome 2 or 1, respectively, with a portion of the FOXO1 or (FKHR) gene on chromosome 13 to generate a PAX3-FOXO1 or PAX7-FOXO1 fusion gene. Each fusion gene is transcribed into a fusion transcript that is in turn translated into a fusion protein. These fusion proteins function as aberrant transcription factors to alter the transcriptional program of the target cell and thereby contribute to the oncogenic phenotype.
In contrast to the translocations found in ARMS, there are no recurrent structural chromosomal changes in ERMS. However, loss of one of the two alleles of many chromosome 11 loci t is a much more frequent occurrence in ERMS than in ARMS.17 This allelic loss localizes to chromosomal region 11p15.5, which is the same region to which the cancer susceptibility gene associated with Beckwith-Wiedemann syndrome and the allelic loss events in other pediatric solid tumors such as Wilms tumor have been localized. In this region, there are multiple genes whose expression is epigenetically regulated in a parent-of-origin-specific fashion by genomic imprinting. A scenario is proposed in which one allele of a tumor suppressor gene in this region is physiologically inactivated by imprinting, and the second allele is removed by the allelic loss event, thereby inactivating both tumor suppressor alleles and promoting an oncogenic effect.
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
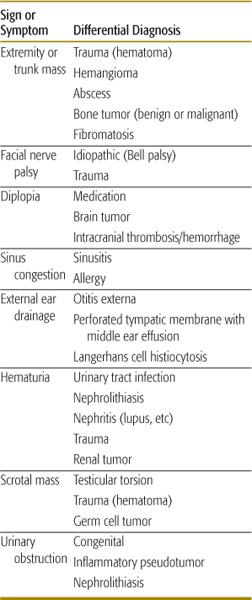
The most common sites of presentation for RMS are the head and neck (including the orbit and parameningeal sites), genitourinary system (including bladder/prostate and paratesticular sites), extremities, and trunk.2-6 The presenting signs and symptoms of RMS are diverse because RMS can originate at any anatomic site (Table 455-1). For example, tumors of the neck or extremity are usually detected as an enlarging mass. Orbital masses can cause proptosis and diplopia. Parameningeal tumors may cause cranial nerve dysfunction (including ophthalmoplegia and facial nerve palsy), external ear drainage, and nasopharyngeal obstruction. Paratesticular tumors present as a scrotal mass. Pelvic or bladder/prostate tumor can present with urinary obstruction and hematuria. The differential diagnosis of a soft tissue mass also varies by anatomic site and presenting symptoms. Perineal and neck masses can result from infectious causes, such as bacterial abscess. Nasopharyngeal masses can appear similar to sinusitis. Benign soft tissue masses, such as hematoma, hemangioma, or fibromatosis, can also mimic RMS.
Table 455-1. Differential Diagnosis by Presenting Symptoms/Signs
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
The investigation of a soft tissue mass in a child or adolescent includes a careful physical examination and diagnostic imaging to define the size, location, and local invasiveness of the primary mass and to detect regional lymphatic metastases. Computerized tomography (CT) and magnetic resonance imaging (MRI) are the most common imaging modalities to assess the local and regional extent of disease. Because of the risk of distant metastatic spread, chest CT, bone scan, and bone marrow aspiration and biopsy are routine staging evaluations for all patients with RMS. Definitive diagnosis is established with a biopsy or resection of the primary mass or biopsy of a metastatic site, depending upon the site’s accessibility and risk of a surgical procedure.
The determination of the presence of RMS within a tissue sample involves several possible diagnostic technologies. Evaluation commences with histopathological evaluation of the sample and detection of a neoplastic population of cells with cytologic and architectural features resembling the ARMS or ERMS patterns described above.15 In some cases, evidence of myogenic differentiation can be seen microscopically by the presence of elongated eosiniphilic “strap-like” cells that may have cross striations or multinucleation. More subtle evidence of myogenic differentiation is provided by immunohistochemical stains directed against muscle-specific proteins such as desmin, or the myogenic transcription factors MyoD and myogenin. Additional evidence of muscle differentiation can also be detected by electron microscopic evaluation of the presence of myofilaments.
To help identify the majority of ARMS cases, tissue samples can also be evaluated for the presence of the 2;13 or 1;13 chromosomal translocations. Though tumor cells can be analyzed by classical cytogenetics, molecular-based testing is faster, more accurate, and less expensive. One popular molecular technology for identifying these translocations is to assay for the PAX3-FOXO1 and PAX7-FOXO1 fusion transcripts in RNA isolated from tissue by the reverse transcription-polymerase chain reaction.18 An alternative technology is to assay for the PAX3-FOXO1 and PAX7-FOXO1 fusion genes in tumor cells or nuclei by fluorescence in situ hybridization. Comparative studies have shown that the results of these two molecular technologies are similar,19 and that ∼60% of ARMS cases are PAX3-FKHR-positive, ∼20% are PAX7-FKHR-positive, and ∼20% are fusion-negative.18
 TREATMENT
TREATMENT
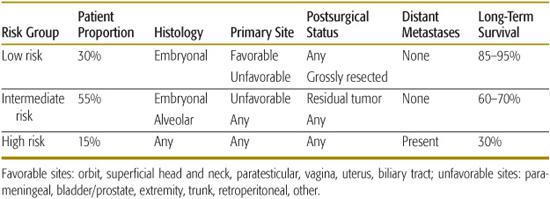
Treatment for RMS is risk adapted, with less radiotherapy and chemotherapy for patients at lower risk for disease recurrence and more therapy for patients at higher risk for recurrence.4-6,20 Factors that most influence success of therapy include presurgical stage (determined by anatomic site, primary tumor size, and regional nodal spread), postsurgical group (defined by the extent of resection), presence of distant metastases, and histologic subtype. These factors establish three risk groups: low-risk, intermediate-risk, and high-risk RMS (Table 455-2).
Table 455-2. Risk Group Stratification
RMS is uniformly treated with systemic chemotherapy. For patients with localized RMS who have initial complete surgical resection, the role of chemotherapy is to treat micrometastatic disease. For initially unresected tumors, chemotherapy can improve the success of local treatment (either radiotherapy or a delayed surgery) to prevent a recurrence at the primary site. Routine use of systemic, multiagent chemotherapy has resulted in impressive improvement in survival rates over the last 30 years. In North America, the most frequently used chemotherapeutic agents are vincristine, dactinomycin, and cyclophosphamide (VAC). Additional agents, including doxorubicin, ifosfamide, and etoposide, are sometimes used to treat patients with metastatic RMS with variable success.4-6,21,22
In addition to chemotherapy, the management of the primary tumor mass includes local radiation therapy and surgical excision.2-6 The selection of either modality depends on the primary site, local invasiveness, and anticipated long-term morbidity of treatment. When feasible without risk of loss of function, initial complete surgical resection is preferred. Completely resected ERMS can be treated with less-intensive chemotherapy, potentially with the omission of radiotherapy. Completely resected ARMS can be treated with lower-dose radiotherapy. Paratesticular primaries and some small, superficial head and extremity primaries are usually treated with surgical excision at diagnosis. Other sites, such as bladder/prostate or vaginal primaries, usually are resectable but with significant long-term morbidity. Parameningeal sites are almost never amenable to resection either at diagnosis or after initial chemotherapy and are usually treated with radiotherapy as the sole modality of local control. Because most children with RMS have immature skeletons, the risk of altered growth after radiotherapy is a consideration in selecting the modality of local treatment. Comparing results from North America (where radiotherapy is used as the primary mode of local control) to those in Europe (where radiotherapy is used more selectively), radiotherapy appears to be more effective in preventing recurrence and improving survival.22,23
 SHORT- AND LONG-TERM COMPLICATIONS
SHORT- AND LONG-TERM COMPLICATIONS
The treatment of RMS is associated with significant short- and long-term toxicity. Multiagent chemotherapy, such as VAC, can cause reversible nausea, alopecia, mucositis, peripheral neuropathy, and bone marrow suppression, with significant risk of life-threatening infection.3 Routine supportive measures can reduce the risk of serious acute side effects. For example, administration of hydration and MESNA can prevent hemorrhagic cystitis that occurs with cyclophosphamide. Prompt use of empiric antibiotics for febrile neutropenia reduces the risk of fatal bacterial infections. Long-term complications of chemotherapy include infertility (due to high cumulative cyclophosphamide doses) and secondary leukemia. Because most children with RMS have not completed linear growth, conventional radiation therapy can impair growth of muscle and bone within the radiotherapy treatment volume. Radiation to the parameningeal site can lead to long-term neurocognitive impairment and growth hormone deficiency and hypothyroidism due to radiotherapy to critical brain structures. Surgery can also cause long-term sequelae, including amputation for an extremity or cystectomy for bladder/prostate RMS.
 PROGNOSIS
PROGNOSIS
For patients without distant metastases, more than 70% of patients will become long-term survivors, with the predicted outcome dependent upon the risk group. The outcome for low-risk patients is particularly favorable, leading to further attempts to limit the extent of treatment, potentially reducing the risk of long-term sequelae. The intermediate-risk patients are treated with moderately intensive chemotherapy and are candidates for clinical trial enrollment to test the addition of new chemotherapeutic agents to improve outcome. In contrast, only 30% of patients with metastatic disease survive long term. High-risk patients are candidates for more innovative therapies to improve their prognosis, including the incorporation of investigational chemotherapeutics. Current research in the treatment of RMS includes the identification of novel agents that target biologic pathways critical to the growth of RMS and functional imaging to determine response to treatment and allow subsequent tailoring of therapy.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


