Retinoblastoma
Rima F. Jubran and Anna T. Meadows
Retinoblastoma (RB) is the most common primary intraocular malignancy of childhood. Incidence rates vary from 1 in 14,000 to 1 in 22,000 live births, and it is estimated that 300 children develop retinoblastoma in the United States annually.1 There is no race or gender predilection. Approximately 80% of children are diagnosed before the age of 3 years. More than half of patients diagnosed in the first year of life have bilateral disease compared with less than 10% of older children.
Retinoblastoma occurs sporadically in the majority of affected children. Of all cases, approximately 60% are unilateral and 25% are bilateral.2 All bilateral cases and 15% of unilateral cases are hereditary. Only 10% to 20% of hereditary cases have an affected parent; therefore, unilateral cases without an affected parent can be determined to be hereditary only with mutation analysis (Fig. 461-1).
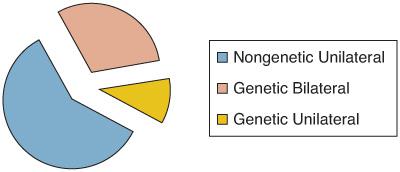
FIGURE 461-1. Retinoblastoma distribution.
 GENETICS
GENETICS
Retinoblastoma (RB) appears to develop only when both alleles of a tumor suppresser gene (RB1) located on 13q14 are absent or defective. In hereditary RB every cell in the body inherits one absent or defective gene. During normal development, a mutagenic “hit” to the remaining normal allele results in the loss of normal cellular growth regulation and tumor development.2 In sporadic, nonhereditary RB, a single retinal cell must receive 2 mutagenic hits to the 13q14 tumor suppressor gene before RB can be expressed. Point mutations of the RB1 gene are the most common events. Variable deletions of the long arm of chromosome 13 are not commonly found in patients with RB and patients who are developmentally delayed should not be presumed to have the chromosome 13q deletion syndrome.3,4
Genetic counseling for patients with retinoblastoma and their families is complex, but a few generalizations can be made. Patients with the hereditary form are at risk for other cancers later in life, including soft tissue and bone sarcomas and melanoma. This risk is increased in patients treated with radiation therapy.5 A survivor of hereditary retinoblastoma has a 50% chance of transmitting the gene to his or her offspring. In patients with unilateral disease, the overall risk of transmitting the gene to offspring is estimated to be 2.5%. With no previous family history, the risk for a sibling is 2% if the affected child has bilateral disease and 1% if the affected child has unilateral disease.6 These empiric risk estimates are based on the possibility of gonadal mosaicism. In addition, it is estimated that 10% of individuals who carry an abnormal RB1 gene do not develop retinoblastoma because the second event did not occur in any cell; however, they are still at risk to develop other cancers during their lifetime.
 PATHOLOGY
PATHOLOGY
Retinoblastoma arises from the inner layer of the retina and usually extends into the vitreous cavity as a white or cream-colored nodular mass. In advanced disease, vitreous seeding and retinal detachment usually occur. The tumor tends to outgrow its blood supply, and patchy necrosis and degeneration with calcium deposition develop.7 Histopathologically, the tumor is composed of small, round blue cells that show variable degrees of photoreceptor differentiation (rosettes and fleurettes).
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
The clinical characteristics of retinoblastoma (RB) depend on the stage of tumor growth at the time of its discovery. When a tumor is small and at the macula, the initial sign may be sensory strabismus. As the tumor grows, leukocoria (white pupil) develops (Fig. 461-2). This is the most common presenting sign of RB, representing 56% to 62% of cases diagnosed.8 When disease is very advanced, presenting symptoms may include pain due to secondary glaucoma, buphthalmos, proptosis, hyphema, and orbital cellulitis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


