Respiratory Distress and Failure
Julio Pérez Fontán
REGULATION OF RESPIRATION
In healthy individuals, respiratory and circulatory functions are linked to tissue metabolic activity by a responsive regulatory system that translates biochemical and neural signals from the tissues into adjustments in cardiac output, vascular tone, and minute ventilation. The purpose of this system is to assure that all the cells in the organism receive a supply of O2 commensurate with their metabolic needs without accumulating excessive amounts of CO2. The system relies both on local circulatory reflexes, which alter the caliber of the supplying blood vessels in accordance with tissue metabolic activity, and on central circulatory and respiratory reflexes, which adjust the pumping function of the heart and the intensity of the respiratory effort in response to changes in the concentration of the respiratory gases in the blood.1
Chemoreceptor reflexes play a singularly important role in the genesis of the manifestations of respiratory disease (Fig. 102-1). Alterations in the blood’s PO2, PCO2, and pH are sensed by specialized chemo-sensitive cells located in the carotid bodies (peripheral chemoreceptors, PO2) and reticular nuclei of the medulla oblongata (central chemoreceptors, PCO2 and pH). These cells relay the information to a medullary neuronal network of premotoneurons, which also receives inputs from mechanical and chemical sensors distributed throughout the lungs, airways, and chest wall. Chemical and mechanical inputs are integrated to define the amount of ventilation needed to sustain adequate gas exchange and the manner in which the respiratory muscles are applied to achieve this ventilation most efficiently. The physiologic basis of respiration is reviewed in more detail in Chapter 503.
RESPIRATORY DISTRESS AND FAILURE
A variety of developmental factors render the infant, and especially the newborn at increased risk for difficulties with respiration. These include immaturity of the neural control of breathing, the small caliber of the airways, the increased flexibility of the rib cage, and a limited respiratory muscle reserve. Furthermore, infants may present with congenital anomalies that impair respiration. Thus, it is not surprising that respiratory dysfunction frequently always occurs when a child becomes critically ill, making it essential that clinicians who care for infants and children recognize the manifestations of respiratory disease. The emergency evaluation of any patient—child or adult—should always begin by assessing the adequacy and characteristics of the patient’s respiratory effort. The phrase respiratory distress denotes an abnormal increase in the effort of the respiratory muscles, typically to overcome an impairment in the mechanical function of the lungs or the chest wall. The increased effort may be noticeable only to the child, who perceives it as shortness of breath (dyspnea), or may also be apparent to an external observer as physical signs. Depending on the severity of the impairment, these usually include a high breathing frequency (tachypnea), nasal flaring (from visible activation of the normally inconspicuous alae nasae muscles), retractions of the intercostal spaces (from the negative swings in pleural pressure generated by the diaphragmatic contractions), and recruitment of accessory muscles (muscles whose primary function is not respiratory but that can help pump air in and out of the lungs). The abdominal muscles (rectus abdominis, oblique muscles, and transversus abdominis) are the most frequently used among the latter. They take advantage of their insertions on the costal cartilages and ribs to stabilize the abdominal wall during inspiration and to accelerate lung emptying during expiration. The scaleni and sternocleidomastoids are also accessory muscles, but, in contrast to the abdominal muscles, they act only to augment rib-cage volume through their distal insertions on the first and second rib and the sternal manubrium, respectively.
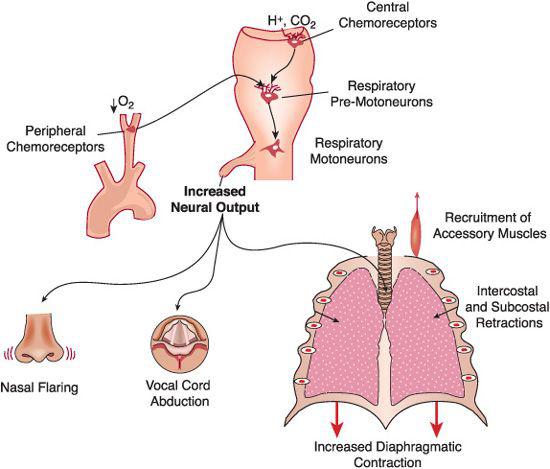
FIGURE 102-1. Genesis of the signs of respiratory distress. Changes in blood-gas tensions are sensed by chemoreceptor cells in the carotid bodies (O2) and in the reticular formation of the medulla oblongata (CO2). The nerve signals originating in the chemoreceptors are integrated and processed by a complex medullary neuronal network that also receives inputs from mechanoreceptors in the lungs and chest wall and from other areas of the brain. Increases in arterial Pco2 and, if sufficiently large, decreases in arterial Po2 sensed by the chemoreceptors activate neural programs that result in the progressive recruitment of a variety of respiratory muscles, as shown in the bottom of the figure. Nasal flaring (from contraction of the dilators of the alae nasae), increased vocal cord abduction, and dilation of the pharyngeal passages during inspiration may not be apparent to the observer. However, alterations in the breathing frequency (usually tachypnea) or intercostal and subcostal retractions (from the subatmospheric pleural pressures generated by the forceful contractions of the diaphragm) are prominent in almost every child with acute respiratory disease.
Increases in the production of CO2 (eg, when metabolic rate is increased by fever or exercise) or in the concentration of hydrogen ions in the blood (eg, metabolic acidosis) also augment neural output to the respiratory muscles, which increases respiratory effort and may mimic respiratory distress. In patients with neuropathy or myopathy signs of respiratory distress, can sometimes be manifested by dyspnea and activation of the respiratory muscles spared by the disease. In patients with severe forms of neuromuscular dysfunction or following the administration of neuromuscular blockers an increased respiratory effort may not be recognized since the respiratory muscles will fail to respond appropriately to the increased respiratory drive.
On occasion, either the neural mechanisms that regulate ventilation fail or, more frequently, the compensatory effort of the respiratory muscles is not sufficient to restore gas exchange to normal, and arterial hypoxemia (an abnormally low arterial PO2) and hypercapnia (an abnormally high PCO2) ensue. This situation, known as respiratory failure, is discussed later in this chapter. It may occur when the effort expended to sustain adequate ventilation cannot be maintained, or when the neural control of breathing is disabled. Respiratory failure is evident when abnormally low respiratory rate (bradypnea), shallow breathing movements, or no respiratory movements at all (apnea) are observed. The breathing control becomes disabled when the central nervous system, especially the brain stem, has suffered a direct injury (eg, cranial trauma, compression by an expanding cerebral tumor or hemorrhage) or is functionally impaired by hypoxia, hypercapnia, or depressants (eg, opioids or barbiturates).
MECHANICAL DYSFUNCTION OF THE LUNGS AND CHEST WALL
During normal respiration forces generated during breathing originate from two types of physical phenomena: one relates to lung inflation; the other relates to expiration and gas flow.
During lung inflation volume-dependent forces arise from elements within the lungs, which oppose inflation and create a tendency for the lungs to recoil. The elastic fibers contained in the alveolar interstitium and in the airways and blood vessels of the lung are a good example. When stretched during inspiration, these fibers behave very much like a rubber band, accumulating energy in their molecular structures, which is then released as they recover their shape during expiration. The more the elastic fibers are stretched, the greater is their tendency to return to their original state or the greater is the overall recoil of the lung. The alveolar gas-liquid interface also acts as a recoil element. When a liquid like the water-based solution lining the alveoli contacts air, water molecules within the liquid phase experience a net push to leave the solution. This push translates into a net force, known as surface tension, which acts to reduce the volume of the alveolus. In the healthy lung, the tendency is relieved greatly by the presence of a lipid monolayer (alveolar surfactant), which separates the water and gas phases and makes the recoil generated from surface tension manageable. When disease or immaturity interferes with surfactant function (see Chapter 54), the increase in surface tension increases the lung recoil, often to levels that prevent adequate inflation. Following inflation, the lung recoils but this recoil is balanced by the outward recoil of the chest wall which distends the lung at rest (Fig. 102-2). Because lung inflation requires the active contraction of the inspiratory muscles (expiration is passive and therefore facilitated by recoil), the physical findings of the diseases in which lung or chest-wall recoil is increased are always more prominent during inspiration than during expiration.
Flow-dependent forces are the result of brief molecular interactions between either the gas flowing through the airways and the airway walls or among tissue components as the lungs change volume. The forces generated by these interactions oppose both inflation and deflation. Just like the velocity of a vehicle determines the fuel consumption incurred in overcoming road and air drag, the magnitude of these forces (often grouped as resistive forces) depends primarily on the speed (or flow rate) at which the lungs inflate or deflate.
Observation of the clinical manifestations of respiratory distress are often sufficient to establish a distinction between two types of disorders that impair respiration: restrictive and obstructive.
 RESTRICTIVE IMPAIRMENTS
RESTRICTIVE IMPAIRMENTS
Restrictive impairments are characterized by an abnormal increase in lung or chest-wall recoil, and therefore they interfere primarily with lung inflation. Examples include pulmonary edema, pneumonitis, interstitial lung disease, and chest-wall deformities that limit lung and chest-wall expansion (see Section 27). In some of these conditions, the presence of fluid (pulmonary edema or pneumonitis), inflammatory cells (pneumonitis), or scar tissue (lung fibrosis) in the interstitial spaces diminishes the ease with which the lung scaffolding can accommodate stretch. In others, the presence of water (advanced pulmonary edema) or exudate (pneumonia) in the alveolar spaces raises surface tension at the gas-liquid interface and reduces the space available for gas in the alveoli. Chest-wall deformities limit lung inflation by making the rib cage more rigid, often asymmetrically. Space-occupying lesions such as pneumothoraces, pleural effusions, and cystic lung anomalies are by definition restrictive, although frequently they distort neighboring airways, thereby creating simultaneous obstructive manifestations.
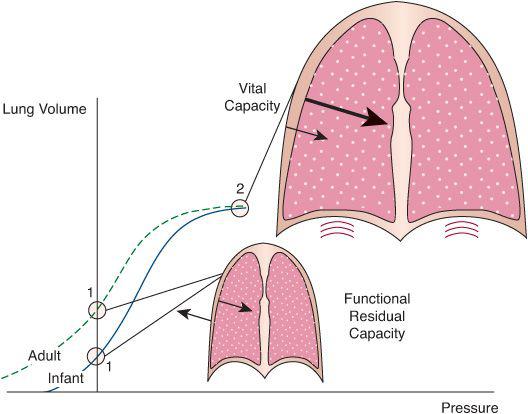
FIGURE 102-2. Idealized comparison of the combined static volume-pressure relationships of the lungs and chest wall in the infant and adult. The relationships shown here are obtained by plotting lung volume against the pressure that the respiratory muscles must generate to maintain the lungs and chest wall at that particular volume. These are determined by measuring lung volume and airway pressure while the lungs are held at a constant volume and the subject relaxes all respiratory muscles against a closed airway or while the lungs are inflated passively in a stepwise manner. The interaction between the lungs and the chest wall is depicted at two points of interest, indicated by circles in the volume-pressure relationships. The first (labeled 1 for both child and adult) is the relaxation volume of the respiratory system, which, under most circumstances, coincides with the functional residual capacity of the lungs. At this volume, the outward elasticity of the chest wall equilibrates with the inward elasticity of the lungs (arrows), and the effective pressure distending the system is zero. Because the immature chest wall has a much smaller tendency to recoil in the outward direction than the mature chest wall, the relaxation volume of the infant’s respiratory system is considerably lower than that of the adult. The second point of interest (labeled 2) is the maximum volume that the lungs can reach during a voluntary inflation (vital capacity). At this volume, both lungs and chest wall generate inward-acting pressures, which are additive, and the diaphragm must generate a great deal of pressure. At any point in the volume-pressure relationship between relaxation volume and vital capacity, the inward-acting forces of the lung predominate and the respiratory muscles must contract to counteract them.
Restrictive impairments increase the effort required to produce an increase in lung volume. This relationship is best represented as a pressure-volume graph, as seen in Figures 102-2 and 503-1. As shown, there is a curvilinear relationship, such that the upper and lower ends of these curves are relatively flat. Large increases in pressure are required for small increases in volume. This type of curve is used to quantitate the severity of restrictive lung disorders.
Healthy individuals breathe in the range of lung volumes corresponding to the steep portion of the relationship, where small changes in pressure promote relatively larger changes in lung volume. However, disease, or sometimes therapeutic interventions, can force the lungs toward one of the flat portions, considerably increasing the effort the respiratory muscles must make to generate a given tidal volume. Mathematically, the volume-pressure relationship is often described by the compliance of the respiratory system, which is defined as the quotient of lung volume and pressure changes (also the slope of the volume-pressure relationship). A reduction in respiratory system compliance for any given lung volume is the fundamental characteristic of restrictive disease. It is important to realize, that because the slope of the volume-pressure relationship changes with lung volume, compliance can be decreased without underlying abnormality of the lung or chest-wall tissue. A child with status asthmaticus, for example, may not have a primary restrictive disease of the lung but will likely have a decreased lung compliance because of overinflated and collapsed alveoli in the lungs that result from the small airway obstruction.
In newborns and infants, incomplete ossification of the rib cage causes the chest wall to be considerably more compliant than in adults.2 This manifests with less outward recoil force from the chest wall, and a predominance of lung recoil in determining lung volumes. Consequently, when the respiratory muscles are fully relaxed, the lungs of the newborn or small infant tend to adopt a smaller volume relative to the lungs of older children or adults. The newborns of most species, including humans, confront this potential mechanical limitation by adopting several strategies to delay expiration, all in an effort to maintain the functional residual capacity of the lungs (the volume of gas contained in the lungs at the end of a normal expiration) above the relaxation volume of the respiratory system.3 These strategies include partially closing the glottis during expiration, prolonging the activation of the diaphragm during part of the expiratory phase, and initiating the next inspiration before exhalation is complete. Neurological dysfunction, anesthesia, and sedation or neuromuscular blockade render these strategies ineffective, making the newborn vulnerable to alveolar collapse and hypoxemia even in the absence of preexisting lung disease.
Clinical Findings in Restrictive Disease
The manifestations of restrictive respiratory disease can be attributed to the mechanical impairment itself, the ensuing increase in the respiratory drive, or the alterations of pulmonary gas exchange that develop if the impairment is not compensated.
Restrictive diseases always increase the inward recoil of the thorax as a whole, independent of whether they primarily affect the lung or chest wall. Therefore, both tidal volume and functional residual capacity tend to decrease (Fig. 102-3). Auscultation of the chest wall demonstrates diminished breath sounds, frequently accompanied by inspiratory and expiratory crackles or rales created by reopening of collapsed air spaces as the effort of the inspiratory muscles exceeds the critical opening pressure. To mitigate the decrease in lung volume, newborns and young infants often close their glottis before exhalation is complete. This may result in a characteristic grunting noise, which is a sign of unstable lung volume and a warning that oxygenation is endangered.4 Chest radiographs are characterized by small sized lung fields and a general appearance of reduced aeration of the lungs in restrictive lung disease. Alveolar collapse may occur in areas of the lung fields where an inefficient cough promotes mucus plugging or the effects of gravity neutralize the net forces that leave alveoli open.
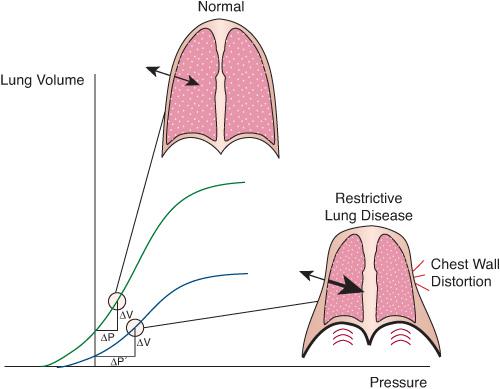
FIGURE 102-3. Effect of restrictive lung disease on the mechanical interaction of lung and chest wall. Restrictive disease displaces the volume-pressure relationship of the lungs downward and to the right. As a result, the decrease in pleural pressure needed to produce a given volume change in the lungs (ΔV) is larger than when the lungs are healthy (ΔP’ versus ΔP). A more negative pleural pressure reduces the outward movement of the chest wall or causes it to move inward in weak areas such as the intercostal, subcostal, and suprasternal spaces, causing chest-wall retractions. The resultant distortion wastes energy, because a substantial portion of the inspiratory muscles’ effort is applied to deform the chest wall without generating effective ventilation of the lungs.
An increase in respiratory drive is triggered by alterations in the blood-gas tensions (chemoreceptor reflexes) or by disease-related changes in the tensile forces within the tissues of the lungs and chest wall (mechanoreceptor reflexes). Patients with restrictive disease typically attempt to increase alveolar ventilation by adopting a pattern of rapid and shallow respirations that compensates for the reduced tidal volume of the shallow breaths. This minimizes the average force that the inspiratory muscles must generate over a given period of time (remember that elastic recoil increases proportionally to lung volume) and the energy cost of breathing.5 In patients with respiratory distress the presence of tachypnea as a prominent sign suggests that the disease is primarily restrictive except in patients with severe respiratory dysfunction, for whom tachypnea may simply be a sign of respiratory muscle fatigue, independent of whether the original impairment is restrictive or obstructive.
Other manifestations of increased respiratory drive are less specific. For example, recruitment of accessory inspiratory muscles such as the scalene and sternocleidomastoid muscles can be detected in children with both restrictive disease and upper-airway obstruction, because in both instances the increased effort is mainly inspiratory. Conversely, recruitment of abdominal muscles during expiration is particularly prominent in patients with intrathoracic obstruction but may also be found in patients with restrictive disease or upper-airway obstruction. In these situations, the contraction of the abdominal muscles helps stabilize the lower rib cage and increases respiratory frequency by accelerating expiration.
Chest-wall retractions are a very common manifestation of respiratory distress in children, but their presence has little value in distinguishing the mechanism responsible for the respiratory distress. The retractions occur during inspiration, when the increased effort of the diaphragm and other inspiratory muscles to overcome lung recoil creates subatmospheric pressures in the pleural space (eFig. 102.1  ). These pressures cause inward deformation of the cartilages and soft tissues of the chest wall, especially at the intercostal, subcostal, and suprasternal areas. The compliant chest wall of the newborn and infant undergoes considerably more deformation for the same degree of lung mechanical impairment than that of older children and adults. In premature and term newborn infants, entire sections of the cartilaginous rib cage can cave during inspiration, adding considerably to the work the diaphragm must perform to ventilate the lungs.6
). These pressures cause inward deformation of the cartilages and soft tissues of the chest wall, especially at the intercostal, subcostal, and suprasternal areas. The compliant chest wall of the newborn and infant undergoes considerably more deformation for the same degree of lung mechanical impairment than that of older children and adults. In premature and term newborn infants, entire sections of the cartilaginous rib cage can cave during inspiration, adding considerably to the work the diaphragm must perform to ventilate the lungs.6
The abnormalities of gas exchange caused by restrictive disease depend on the ability of the respiratory system to maintain alveolar ventilation and on the effectiveness of the local vascular and airway reflex mechanisms that preserve regional ventilation-perfusion ratios within the lungs. These are discussed in more detail later in this chapter.
 OBSTRUCTIVE IMPAIRMENTS
OBSTRUCTIVE IMPAIRMENTS
Obstructive impairments are characterized by an increase in the flow-dependent forces generated through interactions among moving gas and tissue molecules during breathing. The largest portion of these forces originates from friction between air and the relatively narrow airway passages of the infant or child. A smaller proportion originates from molecular interactions within the tissues of the lung and chest wall or in the gas-liquid interface. (These forces may increase in some forms of pulmonary disease, but their contribution to the overall mechanical dysfunction of the respiratory system is not well characterized.)
From a mechanical perspective, obstructive impairments have two distinguishing characteristics. First, any work that the respiratory muscles perform to overcome the obstruction is ultimately dissipated as heat (unlike restrictive disease, where the work done by elastic forces is stored during inspiration and used to facilitate expiration). Because energy that is transformed into heat leaves the system immediately, the relationship obtained by plotting the pressure generated by the respiratory muscles against the volume change of the lungs follows a different trajectory during inspiration and expiration (indicating that pressure diminishes even if volume is the same), a phenomenon known as hysteresis. Second, the magnitude of the energy losses incurred at the obstruction (and thus the pressure necessary to compensate these losses) depends on the gas’s velocity. Gas velocity, in turn, is determined by the gas-flow rate and thus depends on the speed with which the lungs inflate and deflate.
The pressure that the respiratory muscles generate to overcome obstruction increases as respiratory rate rises. However, the exact terms of the relationship between pressure and gas flow are defined by the manner in which the gas molecules travel in the flow stream. During normal breathing, flow in most airways adopts a layered or laminar pattern, whereby the molecules in the central layers travel faster than those in the outer layers, which are slowed down by the drag of the airway wall. Under such conditions, frictional pressure losses are proportional to the length of the airways and the viscosity of the gas and are inversely related to the fourth power of the airway’s radius (Poiseuille’s law). The laminar flow pattern is disrupted when flow velocity increases, or when irregularities develop in the airway wall. Gas molecules start to move randomly, in a pattern known as turbulent flow, which dissipates more energy than laminar flow and therefore requires increased respiratory muscle effort to move less air. Furthermore, because pressure losses during turbulence are caused by molecular collisions, the magnitude of these losses depends on gas density rather than on gas viscosity. This is one of the reasons why patients with obstructive airway disease may improve when breathing high concentrations of helium, which has a lower density than air or O2 (even though its viscosity is slightly greater).
In normal airways, viscous friction predominates. The loss of energy dissipated as heat causes the pressure inside the airways to decline gradually in the direction of flow. For this reason, alveolar pressure is always lower (or more negative) than the pressure measured elsewhere in the airway tree during inspiration and is higher (or less negative) during expiration. When there is a discrete obstruction, the gradual decline in pressure turns into a sudden step. Gradual or sudden, the changes in the pressure inside the airways have important effects on the size of the airway lumen. Indeed, unlike rigid pipes, airways vary their caliber depending on the net balance of pressures acting on their inside and outside wall surfaces (known as airway transmural pressure). A positive transmural pressure (inside pressure is greater than outside) increases airway caliber; a negative transmural pressure (outside pressure greater than inside pressure) decreases airway caliber and, depending on the rigidity of the airway wall, may cause the airway lumen to collapse altogether.
Airway obstruction can reduce the pressure distending the inside of the wall by two different mechanisms. First, as discussed earlier, increased friction and, when present, turbulence cause a step reduction in pressure downstream from the obstruction. Second, to accommodate flow as the airway narrows, the gas molecules must accelerate and gain kinetic energy. This kinetic energy gain can only occur with a reduction of the potential energy stored from the force distending the airway wall. As the velocity increases, the pressure on the wall decreases and the airway caliber decreases.
These effects of the obstruction on airway caliber further diminish the amount of flow the obstructed airway can accommodate. To compensate, the respiratory muscles need to contract during expiration to force increased flow through the obstruction. However, there is a limit at which the increase in driving pressure is offset by a further reduction in airway caliber caused by the increase in pressure applied on the airway’s outside surface (viscous flow limitation) or by the decrease in the inside pressure as flow needs to be accelerated even more (wave-speed flow limitation). The maximum flow achievable under those circumstances depends on the location and severity of the obstruction and on the rigidity of the airway wall. Flow limitation is common in diseased airways and contributes substantially to the manifestations of airway obstruction.7
Clinical Findings in Obstructive Disease
The manifestations of obstructive respiratory disease depend on the hierarchy of the obstructed airways and the severity of the obstruction. Obstructions of the larynx and trachea affect gas flow to both lungs and therefore represent a much greater threat than localized bronchial obstructions.
Because gas flow defines the rate at which the lungs change volume, some delay in inspiration, expiration, or both is always detectable. The delay lengthens the total duration of each breath. Thus, tachypnea is a less prominent sign of respiratory distress (when not absent altogether) in patients with airway obstruction than in patients with restrictive disease.
Determining whether the impediment to gas flow is predominantly inspiratory or expiratory helps considerably in the differential diagnosis of airway obstruction. As a rule, obstructions of the extrathoracic airway (nose, pharynx, larynx, and cervical segment of the trachea) are exacerbated during inspiration; obstructions of the intrathoracic airway (thoracic segment of the trachea, bronchi, and bronchioles) are exacerbated during expiration. These are an exaggeration of the normal fluctuations of the airway caliber during unobstructed breathing. They result from differences in how breathing affects the transmural pressure of the extra- and intrathoracic airways (eFig. 102.1  ).
).
The clinical signs of extrathoracic airway obstruction are more prominent during inspiration, which is prolonged and obviously more strenuous than expiration. A high-pitched noise, or stridor, produced by the vibration of the airway mucosa at the level of the obstruction can usually be heard without the help of a stethoscope. When the obstruction is severe, a child may sit in a forward-leaning position, resting on both arms to facilitate the use of the neck accessory muscles during inspiration.
The physical findings of intrathoracic obstruction are more prominent during expiration. The expiratory phase is prolonged, and wheezing (a whistling noise produced by high-frequency vibrations of the airway wall and gas column) can be auscultated over the obstructed area or, if the obstruction is diffuse (eg, asthma), over the entire lung fields. Inspiration is less affected than expiration, because the obstruction is partially relieved by the decrease in pleural pressure during this phase of the breathing cycle. The preferential impairment of expiratory gas flow may not be compensated entirely by the prolonged expiratory phase. If so, the alveolar spaces subtended by the obstructed airways do not empty entirely before the next inspiration starts, and the volume of the affected alveoli at end-expiration increases (a condition easily identified on chest radiographs that is frequently referred to as gas trapping). However, the alveolar volume increase is limited by the effects of distention on lung recoil, which dictate an equilibrium whereby the increased recoil limits tidal volume and accelerates exhalation enough to compensate for the low expiratory flow.
RESPIRATORY FAILURE
The main functions of the respiratory system are to replenish the venous blood’s content of O2 while removing its excess CO2. Respiratory failure describes the inability to carry out this function commensurate with the needs of the organism. This definition may be misleading. For example, to state that respiratory failure exists when the partial tensions of O2 (PO2) and CO2 (PCO2) in the arterial blood remain persistently outside the range found in normal humans (values of PO2 < 50 mm Hg or PCO2 > 45 mm Hg are often cited). However, this definition ignores the fact that breathing O2-enriched gas can by itself restore the arterial PO2 to the normal range, even though the gas-exchanging mechanism is faulty and may not sustain the needs of the individual while breathing air. Conversely, the same definition would categorize children with congenital cyanotic heart disease (who are hypoxemic) or with diuretic-induced metabolic alkalosis (who, as a compensatory mechanism, are hypercapnic) as suffering from respiratory failure, even though their respiratory system may be perfectly functional.
For these reasons, rather than detecting specific aberrations in blood-gas content, the clinician’s primary concern should be whether the respiratory system can support metabolic demands under all the circumstances the patient is likely to encounter. Fever or exercise often unveil otherwise compensated anomalies of gas exchange by adding to the ventilatory load of the respiratory muscles or by increasing demand for pulmonary blood flow. Hypoxemia and hyper-capnia interfere most noticeably with the function of the central nervous and cardiovascular systems. Agitation, somnolence, apathy, combativeness, or even stupor in an infant or child with respiratory distress or decreased respiratory effort should at least prompt the administration of O2, even if blood-gas analysis is unavailable. Increasing tachycardia, arterial hypertension, or, by way of progression, decreased perfusion, arterial hypotension, and bradycardia are worrisome signs in a patient with a respiratory derangement and, under most circumstances, constitute an immediate indication to institute artificial ventilatory support.
 DISRUPTION OF GAS EXCHANGE
DISRUPTION OF GAS EXCHANGE
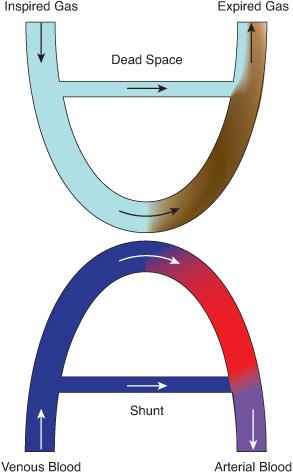
Understanding the mechanisms that lead to blood-gas aberrations in infants and children with respiratory failure is essential to interpreting clinical information and to planning treatment.8 Basic to such understanding is the notion that the exchange of gases between the inspired gas and the blood in the lungs produces two products: the expired gas and the arterial blood (Fig. 102-4). Each of these products is a composite of the contributions of millions of alveolar-capillary units, weighted by the amount of oxygen or carbon dioxide in blood or gas from each unit. In every one of these units, and in the lung as a whole, the gas contents of the alveolar gas and the capillary blood are linked reciprocally by relatively simple laws that establish a framework for understanding the gas-exchanging process and its abnormalities.
Oxygen and CO2 are highly diffusible gases. Diffusion impairments across the alveolar-capillary membrane play little role in the genesis of hypoxemia or hypercapnia in children with respiratory disease. Accordingly, it is safe to assume that the PO2 and PCO2 of the blood exiting a given pulmonary capillary reflects the PO2 and PCO2 of the gas contained in the corresponding alveolus. The composition of the alveolar gas is determined by the rate at which gases are exchanged across the alveolar-capillary membrane (diffusion and perfusion), and the turnover rate of fresh air entering the alveoli (ventilation). Mathematical modeling of the relationship between alveolar PO2, ventilation, and O2 uptake is relatively complicated,9 because O2 is present in both the inspired and the expired gas. In contrast, modeling of the relationship between PCO2, ventilation, and CO2 elimination is relatively simple, because the inspired gas does not contain CO2. For the whole lung, the average alveolar PCO2 is directly proportional to the amount of CO2 produced by the body and is inversely proportional to the volume of gas that participates in alveolar gas exchange per unit of time. The latter is known as alveolar ventilation (as opposed to minute ventilation, which is the total amount of gas that exits the lungs per unit of time or the product of tidal volume by breathing frequency).
The dead space, or wasted ventilation (Fig. 102-4) is the portion of the total minute ventilation that does not participate in CO2 exchange. This generally represents the gas that is expired from the lungs that remains in the large airways during expiration, until re-inspired into the alveoli during the next inspiration. Since this gas still contains CO2, it does not contribute to elimination of CO2 from the lungs and therefore is shown in Figure 102-4 as not contributing to CO2 gas exchange. Disorders that increase the dead space usually do not cause hypercapnia because the individual is able to increase minute ventilation sufficiently to restore alveolar ventilation to the needed level to maintain CO2 within a normal range (as occurs when breathing through a short piece of tubing). Alveolar CO2 levels determine the pulmonary arterial blood CO2 concentrations. 
Arterial blood gas concentrations can be conceptualized as the product of two idealized components. One consists of systemic venous blood that bypasses (or is shunted away from) the alveoli and hence does not participate in gas exchange. The other arises from blood that undergoes perfect exchange with alveolar gas. The mixture of these two components determines the actual arterial blood gas concentrations.
For simplicity, the source of the blood that bypasses alveoli (or venous admixture in the physiological parlance) has conventionally been viewed as arising from several discrete pathways. First, true anatomic shunt follows anatomic communications between the venous and arterial side of the circulation. Some of these communications are found in normal individuals (eg, thebesian veins, which connect the coronary circulation to the left ventricle, or the bronchial vessels, which direct venous blood into the pulmonary veins); others result from cardiac malformations (see Chapter 484) or lung disease resulting in alveolar collapse or consolidation (ventilation-perfusion ratio of 0). Second, diffusion defects, whether caused by true alterations of the alveolar capillary membrane or, more frequently, by blood being forced through the capillaries at a rate that does not permit equilibration with the alveolar gas, reduce the end-capillary PO2 and have an effect similar to mixing venous blood with fully oxygenated blood. Finally, ventilation-perfusion inequality results primarily from incomplete oxygenation of blood circulating through lung units with a low ventilation-perfusion ratio. Diffusion abnormalities and ventilation-perfusion inequality create only a virtual shunt, which decreases if the inspired oxygen concentration is increased. In contrast, when a true anatomic shunt exists, O2 administration can only increase the oxygen content of the arterial blood by raising the volume of O2 dissolved in the pulmonary capillary blood; thus, the calculated size of the shunt is affected minimally by O2 administration.
Ventilation-perfusion inequality is by far the most common mechanism of hypoxemia and hypercapnia,10 both in children and adults with respiratory disease (Fig. 102-5). Ventilation-perfusion differences are a natural consequence of the parallel organization of the bronchial and arterial networks of the lungs, which permits an infinite combination of ventilation-perfusion ratios to coexist in the same lung. Gravity causes a certain degree of ventilation-perfusion inequality in normal lungs by directing a larger share of blood flow to dependent areas. Bronchial obstruction, consolidation or collapse of alveolar spaces, and abnormalities in pulmonary vascular function greatly exaggerate this inequality. The cause of hypoxemia in ventilation-perfusion inequality lies primarily with the alveolar-capillary units that have a low ventilation-perfusion ratio. Because renewal of the alveolar gas through ventilation cannot keep up with O2 uptake by the blood, these units have a low alveolar PO2, usually in a range where the O2-hemoglobin dissociation curve is steep. As a result, the end-capillary blood is not fully loaded with O2, and when mixed with blood from other units, it creates a substantial venous admixture. Units with high ventilation-perfusion ratios have a high alveolar PO2. However, these units cannot compensate for the venous admixture caused by units with low ventilation-perfusion ratios, because at high PO2 levels, the O2-hemoglobin dissociation curve is flat, and the blood cannot increase its O2 content substantially.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


