Respiratory Chain Disorders
Salvatore DiMauro
DISORDERS OF THE MITOCHONDRIAL RESPIRATORY CHAIN
The respiratory chain (RC) is the terminal pathway of mitochondrial metabolism, where most energy is produced as adenosine triphosphate (ATP). It is also the only metabolic pathway under dual genetic control: Of the approximately 80 subunits of the RC, 13 are encoded by mitochondrial DNA (mtDNA) and the rest by nuclear DNA (nDNA). Defects of the RC cause an extremely heterogeneous group of disorders that affect both children and adults, often involving multiple tissues and resulting in characteristic syndromes but sometimes affecting single tissues. Disorders due to mutations in mtDNA are especially challenging for the clinician, because the rules of mitochondrial genetics make for intrafamilial variability, including often elusive maternal inheritance, syndromic or nonsyndromic multisystem involvement, and variably severe laboratory abnormalities. Disorders due to mutations in nDNA are inherited as Mendelian traits and include “direct hits,” or mutations directly affecting subunits of the RC; “indirect hits,” or mutations in proteins needed for the proper assembly of individual RC complexes; and defects of integenomic signaling, such as mutations in proteins needed for the maintenance of mtDNA (translation, replication, repair). The central disorder of pediatric interest is Leigh syndrome (LS), which reflects the consequences of impaired energy metabolism on the developing brain and is characterized clinically by psychomotor regression and signs of brain-stem dysfunction; radiologically it is characterized by bilateral, symmetrical lesions in the basal ganglia and the brain stem. LS is associated with both mtDNA- and nDNA-related disorders and with defects of pyruvate metabolism (see Chapter 159).
HISTORY
The first pathogenic mutations in mtDNA were reported 1988, when Holt and colleagues described large-scale deletions in patients with mitochondrial myopathies,1 and Wallace and colleagues described a point mutation in the gene encoding subunit 4 of complex I (ND4) in a family with Leber’s hereditary optic neuropathy (LHON).2 However, these two papers opened up a veritable Pandora’s box: the “morbidity map” of mtDNA has gone from the one-point mutation of 1988 to over 200 pathogenic point mutations in 2008 (Fig. 158-1).3 In 1995, Bourgeron and colleagues described the first “direct hit” mutation in nDNA in two sisters with LS and complex II deficiency4; the first “indirect hit” also caused LS and was due to mutations in SURF1, a protein needed to assemble cytochrome c oxidase (COX, complex IV of the respiratory chain [RC])5,6; two papers, one in 1989 the other in 1991, suggested that mutations in nDNA-encoded maintenance proteins were responsible for multiple mtDNA deletions in patients with autosomal dominant progressive external ophthalmoplegia (PEO)7 and with mtDNA depletion in two infants with myopathic or hepatocerebral presentations.8 Multiple specific nuclear genes have since been associated with defects of intergenomic communications.9
MITOCHONDRIAL GENETICS
Given the importance of mtDNA-related diseases, all practicing pediatricians ought to be familiar with the rules of mitochondrial genetics, which differ from those of Mendelian genetics in the following ways:
1. Heteroplasmy and threshold effect. Each cell contains hundreds or thousands of mtDNA copies, which, at cell division, distribute randomly among daughter cells. In normal tissues, all mtDNA molecules are identical (homoplasmy). Most deleterious mutations of mtDNA usually affect some but not all mtDNAs within a cell, a tissue, or an individual (heteroplasmy), and the clinical expression of a pathogenic mtDNA mutation is largely determined by the relative proportion of normal and mutant genomes in different tissues. A minimum critical number of mutant mtDNAs is required to cause mitochondrial dysfunction in a particular organ or tissue and mitochondrial disease in an individual (threshold effect).
2. Mitotic segregation. At cell division, the proportion of mutant mtDNAs in daughter cells may shift, and the phenotype may change accordingly. This phenomenon, called mitotic segregation, explains how certain patients with mtDNA-related disorders may actually shift from one clinical phenotype to another as they grow older.
3. Maternal inheritance. At fertilization, all mtDNA derives from the oocyte. Therefore, the mode of transmission of mtDNA and of mtDNA point mutations (single deletions of mtDNA are usually sporadic events) differs from Mendelian inheritance. A mother carrying an mtDNA point mutation will pass it on to all her children (males and females), but only her daughters will transmit it to their progeny. A disease expressed in both sexes but with no evidence of paternal transmission is strongly suggestive of an mtDNA point mutation.
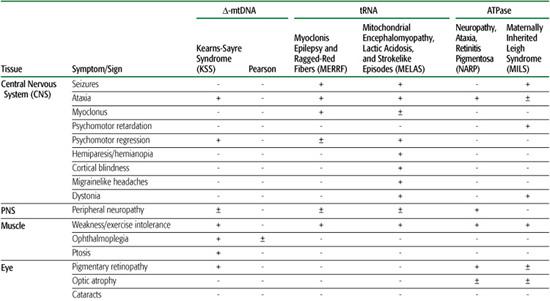
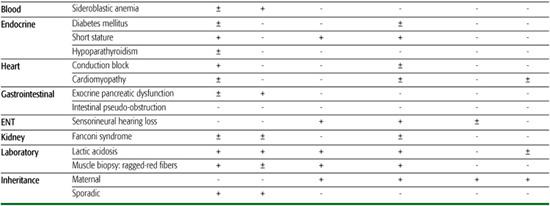
Over 200 point mutations and innumerable large-scale rearrangements have been associated with a bewildering variety of diseases (Figs.158-1 and 158-2). This is not surprising when one considers that mitochondria are ubiquitous organelles and that all human tissues, in isolation or in various combinations, can be affected by mtDNA mutations.10 This concept is illustrated in Table 158-1, which summarizes the symptoms and signs described in mitochondrial encephalomyopathies due to single rearrangements (Kearns-Sayre syndrome [KSS], Pearson syndrome [PS], and progressive external ophthalmoplegia [PEO]); due to point mutations in genes affecting protein synthesis in toto (mitochondrial en-cephalomyopathy, lactic acidosis, and strokelike episodes [MELAS]; myoclonus epilepsy and ragged-red fibers [MERRF]); and due to mutations affecting a protein-coding gene (neuropathy, ataxia, retinitis pigmentosa [NARP] and maternally inherited Leigh syndrome [MILS]). This table highlights the clinical features of the most common syndromes, which are difficult to miss in typical patients. However, it also serves to remind the astute clinician that any combination of these symptoms and signs should raise the suspicion of an mtDNA-related disorder.
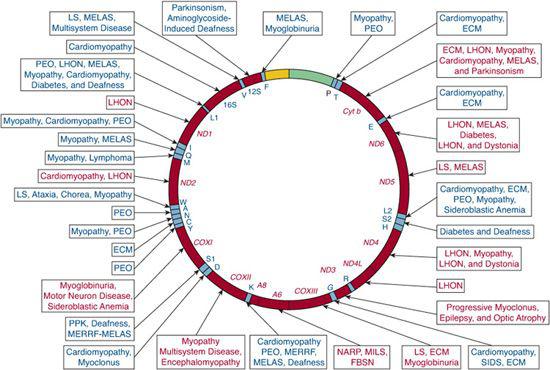
FIGURE 158-1. Morbidity map of the human genome. Differently shaded areas in the 15.5 kb mtDNA map represent the protein-coding genes for the seven subunits of complex I (ND), three subunits of complex IV (COX), cytochrome b (Cyt b), two subunits of ATP synthetase (ATPase 6 and 8), 12S and 16S rRNA, and the 22 trNAs. One-letter codes represent the corresponding amino acids. FBSN, familial bilateral striatal necrosis; KSS, Kearns-Sayre syndrome; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes; MERRF, myoclonic epilepsy with ragged-red fibers; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia, retinitis pigmentosa; PEO, progressive external ophthalmoplegia.
CLINICAL PRESENTATIONS
 DISORDERS DUE TO MTDNA MUTATIONS
DISORDERS DUE TO MTDNA MUTATIONS
Among the maternally inherited encephalomyo-pathies, four syndromes are more common. The first is MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes), which usually presents in children or young adults after normal early development. Symptoms include recurrent vomiting; migrainelike headaches; and strokelike episodes causing cortical blindness, hemiparesis, or hemianopia. MRI of the brain shows “infarcts” that do not correspond to the distribution of major vessels, raising the question of whether the strokes are vascular or metabolic in nature. The most common mtDNA mutation is A3243G in the tRNALeu(UUR) gene, but about a dozen other mutations have been associated with MELAS.11
The second syndrome is MERRF (myoclonus epilepsy with ragged-red fibers), characterized by myoclonus, seizures, mitochondrial myopathy, and cerebellar ataxia. Less common signs include dementia, hearing loss, peripheral neuropathy, and multiple lipomas. The typical mtDNA mutation in MERRF is A8344G in the tRNALys gene, but other mutations in the same gene have been reported.11
The third syndrome comes in two subtypes. The first is NARP (neuropathy, ataxia, retinitis pigmentosa), which usually affects young adults and causes retinitis pigmentosa, dementia, seizures, ataxia, proximal weakness, and sensory neuropathy. The second is a maternally inherited form of Leigh syndrome (MILS). NARP and MILS can affect maternal relatives in the same family.
The fourth syndrome, Leber’s hereditary optic neuropathy (LHON), is characterized by acute or subacute loss of vision in young adults, more frequently males, due to bilateral optic atrophy. Mutations in three genes of complex I (ND genes) have been associated with LHON: G11778A in ND4, G3460A in ND1, and T14484C in ND6.12
Three sporadic conditions are associated with mtDNA single deletions: Kearns-Sayre syndrome (KSS), progressive external ophthalmoplegia (PEO), and Pearson syndrome (PS). KSS is a multisystem disorder with onset before age 20; symptoms include impaired eye movements (PEO), pigmentary retinopathy, and heart block. Frequent additional signs include ataxia, dementia, and endocrine problems (diabetes mellitus, short stature, hypoparathyroidism). Lactic acidosis and markedly elevated cerebrospinal fluid (CSF) protein (over 100 mg/dL) are typical laboratory abnormalities.13
PEO is a relatively benign disorder characterized by ptosis, PEO, and proximal myopathy. Sideroblastic anemia and exocrine pancreatic dysfunction characterize Pearson syndrome, a usually fatal disorder of infancy.
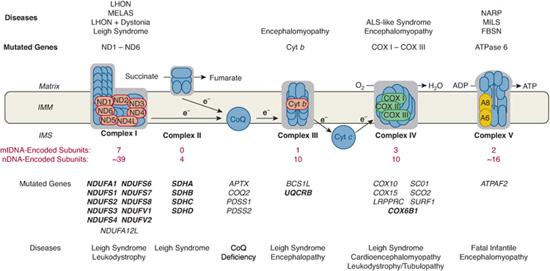
FIGURE 158-2. Schematic representation of the mitochondrial respiratory chain, showing nDNA-encoded subunits (blue) and mtDNA-encoded subunits in different colors. Protons are pumped from the matrix to the intermembrane space (IMM) through complexes I, III, and IV and are pumped back to the matrix through complex V to produce ATP. Coenzyme Q (CoQ) and cytochrome c (Cyt c) are electron (e-) transfer carriers. Diseases due to mutations in mtDNA (above the CR) or to mutations in nDNA (below the CR) are listed according to the correspondingly affected RC complex. Mutant genes shown in bold encode subunits of RC complexes (“direct hits”); mutant genes shown in plain font encode assembly factors. ALS, amyotropic lateral sclerosis; FBSN, familial bilateral striatal necrosis; LHON, Leber hereditary optic neuroretin-opathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MILS, maternally inherited form of Leigh syndrome; NARP, neurogenic ataxia retinitis pigmentosa; SDH, succinate dehydrogenase; TCA, tricarboxylic acid.
Two mtDNA genes encoding subunits of complex I (ND3 and ND5) have to be considered when confronted with patients with Leigh syndrome (LS) or MELAS, even if maternal inheritance is missing or not obvious, if ragged-red fibers are not abundant, and if lactic acidosis is not severe.14
It is often stated that any patient having multiple organ involvement and evidence of maternal inheritance should be suspected of harboring a pathogenic mtDNA mutation until proven otherwise. While this rule of thumb has some practical value, it is also important to remember that the reverse is not true—that is, patients with involvement of a single tissue and no evidence of maternal inheritance can still have pathogenic mutations of mtDNA. This is especially true for skeletal muscle. We have mentioned previously how isolated myopathy with PEO can be due to single large-scale mtDNA deletions. Isolated myopathy without PEO has been associated with point mutations in several tRNA genes.15 In addition, exercise intolerance, myalgia, and myo-globinuria can be the sole presentation of respiratory chain defects due to mutations in protein-coding genes.15
 DISORDERS DUE TO MUTATIONS IN NDNA
DISORDERS DUE TO MUTATIONS IN NDNA
“Direct hits” refer to mutations in genes encoding subunits of the respiratory chain. Until very recently, only pathogenic mutations in subunit A of complex II and in a handful of complex I subunits have been associated with disease, usually LS and leukoencephalopathy (Fig. 158-2). A mutation in the ubiquitously expressed COX6b1 subunit has now been associated with cystic leukoencephalopathy, the first direct hit affecting complex IV.16
“Indirect hits” are mutations that alter proteins that are not subunits of the respiratory chain (RC) but that are indispensable for the correct assembly of RC complexes. These have been described for all complexes except complex II (Fig. 158-2). They cause a variety of multisystemic syndromes, often characterized by encephalopathy (Leigh syndrome [LS] or LS-like) and the selective involvement of another organ or tissue, which can offer a diagnostic clue (eg, the cardiopa-thy in patients with SCO2 mutations). For practitioners, mutations in the SURF1 gene are especially important, because they are the most common causes of COX-deficient LS, which in turn is one of the most common forms of LS. Neuroradiological LS-like lesions are also present in infants with congenital lactic acidosis; renal tubulopathy; liver failure with hepatosiderosis; and encephalopathy with psychomotor retardation, a devastating syndrome labeled GRACILE (growth retardation, aminoaciduria, cholestasis, iron overload, lactacidosis, and early death) due to mutations in an assembly protein (BCS1L) for complex III (Fig. 158-2).
A recent addition to this group of disorders is coenzyme Q10 (CoQ10) deficiency, which can cause four major autosomal recessive syndromes. The first is a predominantly myopathic form, with exercise intolerance and recurrent myoglobinuria, as well as central nervous system involvement, with seizures, ataxia, or mental retardation. The second is a predominantly ataxic form, mimicking spinoc-erebellar ataxia, with prominent cerebellar atrophy and a variety of inconsistently associated features, including seizures, mental retardation, pyramidal signs, and peripheral neuropathy. The third syndrome is a pure myopathy, with exercise intolerance and proximal limb weakness. The fourth presentation is a mitochondrial encephalomyopathy associated with nephrosis: This association is an important clue to the correct diagnosis, because other RC disorders are often associated with renal tubular dysfunction but rarely with glomerular disease. As has long been suspected, mutations in several genes directly or indirectly involved in CoQ10 biosynthesis have been identified,17-19 and more will undoubtedly be discovered. Diagnosing CoQ10 deficiency has practical importance, because many patients respond, some dramatically, to CoQ10 supplementation.
Table 158-1. Clinical Features in Mitochondrial Disease Due to mtDNA Mutations
 DISORDERS DUE TO DEFECTS IN INTERGENOMIC SIGNALING
DISORDERS DUE TO DEFECTS IN INTERGENOMIC SIGNALING
In these disorders, mutations in nuclear genes cause qualitatively (multiple deletions) or quantitatively (depletion) alterations of mtDNA. Multiple deletions and depletion often coexist, because they are commonly due to altered control of the intramitochondrial nucleotide pool, which is essential for maintaining mtDNA.
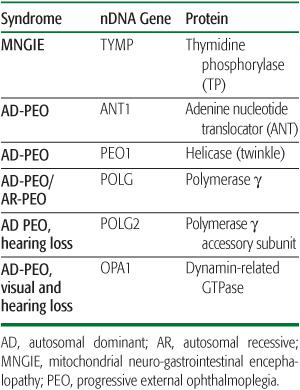
The most common clinical feature of patients with multiple mtDNA deletions in skeletal muscle is progressive external ophthalmoplegia (PEO), which can be inherited as an autosomal dominant or recessive trait (Table 158-2). Autosomal dominant PEO has been associated with mutations in four genes that encode proteins involved in mitochondrial nucleotide metabolism: ANT1, encoding an isoform of the adenine nucleotide transporter; PEO1 (formerly known as Twinkle), encoding a helicase; and POLG, encoding the mitochondrial g polymerase. Autosomal dominant PEO with visual and hearing loss can also be due to mutations in OPA1, which encodes a dynamin-related GTPase essential for mitochondrial motility and is more often associated with dominant optic atrophy (DOA), the Mendelian counterpart of the maternally inherited Leber hereditary optic neuropathy (see above).
Mutations in POLG are especially interesting for many reasons. First, they appear to be the most common causes of Mendelian PEO. Second, they can cause both dominant and recessive forms of PEO. Third, they have been associated with a striking variety of clinical features, including sensory ataxic neuropathy, dysarthria, and opthalmoplegia (SANDO), mitochondrial recessive ataxic syndrome (MIRAS), deafness, hypogonadism, seizures, myoclonus, affective disorders, and gastrointestinal dysmotility. Fourth, and crucially important for pediatricians, POLG mutations are the most common causes of Alpers syndrome, a disorder of childhood characterized by liver insufficiency and gray matter involvement (poliodystrophy) and accompanied by mtDNA depletion (see below). This clinical heterogeneity is largely explained by the complexity of the enzyme, which comprises an exonuclease domain with proofreading function, and a polymerase domain with mtDNA-replicating function, connected by a “linker” domain.20 A fifth noteworthy feature of POLG mutations is that they can also affect the gene (POLG2) encoding the accessory rather than the catalytic subunit and can cause autosomal recessive PEO.21
Table 158-2. Syndromes, Genes, and Proteins Associated with Multiple mtDNA Deletions
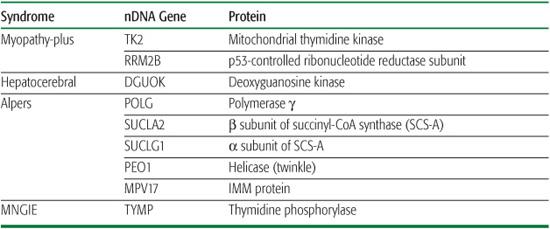
A special autosomal recessive form of PEO that combines multiple deletions and depletion of mtDNA in muscle is MNGIE (mitochondrial neurogastrointestinal encephalomyopathy), a disorder of young adults characterized by PEO, peripheral neuropathy, leukoencephalopathy, and severe gastrointestinal dysmotility leading to cachexia and early death. The gene (TYMP) responsible for MNGIE encodes the enzyme thymidine phosphorylase (TP): although extra-mitochondrial, mutated TP alters the homeo-stasis of the intramitochondrial nucleotide pool.22
Two main syndromes have been associated with mtDNA depletion, one dominated by my-opathy and the other by liver and brain dysfunction.9 Myopathic mtDNA depletion can be fatal in infancy or later in childhood due to respiratory failure, but it may also affect the CNS and mimic spinal muscular atrophy.23 The hepatoce-rebral form of mtDNA depletion usually manifests in infancy or early childhood and has been attributed to mutations in POLG (see above), DGUOK (encoding deoxyguanosine kinase), SUCLA2 (encoding the b subunit of succinyl-CoA-synthase [SCS-A]), SUCLG1 (encodingthe a subunit of SCS-A), or RRM2B (encoding a p53-controlled ribonucleotide reductase subunit [p53R2]). Autosomal recessive hepatocerebral syndrome was also attributed to mutations in two genes that are not directly involved in nucleotide metabolism: MPV17 (encoding an inner mitochondrial membrane protein)24 and PEO125 (mutations in this gene can also cause autosomal dominant PEO and multiple mtDNA deletions, as described above). A homozygous mutation (R50Q) in MPV17 causes a distinctive severe disorder dominated by peripheral neuropathy and liver failure in children of the Navajo Nation (Navajo neurohepatopathy, NNH).26
Defects of mtDNA translation have been associated with mutations in various nucleus-encoded factors needed for mitochondrial protein synthesis, including mitoribosomal proteins (MRPS16; MRPS22, resulting in multisystemic disease); mRNA-specific translation factors (LRPPRC, resulting in Leigh syndrome, Canadian type); general translation factors (EFG1, resulting in hepatocerebral syndrome; TSFM, resulting in encephalomyopathy or cardiomyopathy); tRNA processing and base-modification enzymes (PUS1, resulting in myopathy, lactic acidosis, and sideroblastic anemia [MLASA]); and aminoacyl-tRNA synthetases (Arg-tRNA synthetase, resulting in pontocerebellar hypoplasia; Asp-tRNA synthetase, resulting in leukoencephalopathy; Gly-tRNA synthetase, resulting in Charcot-Marie-Tooth type 2 [CMT2], or in spinal muscular atrophy type V [SMA V]).27 As rare as these disorders are—at least until now—they should be considered in the differential diagnosis of more common pediatric neurology disorders, such as LS, leukodystrophy, and hepatocerebral syndrome.
 DEFECTS OF THE LIPID MILIEU
DEFECTS OF THE LIPID MILIEU
The inner mitochondrial membrane (IMM) is essential for respiratory chain (RC) function. Thus, alterations of the phospholipid composition of the MIM may result in RC defects. This appears to be the case of Barth syndrome, an X-linked disorder of childhood manifesting as myopathy, cardiomyopathy, leucopenia, and growth retardation. The gene responsible for Barth syndrome (initially dubbed G4.5 and now called TAZ) encodes a family of proteins called tafazzins, which share conserved regions with acyltransferases of diverse organisms. It has been documented in Barth syndrome that mutant tafazzins lead to impaired or altered synthesis of cardiolipin, the major phospho-lipids component of the IMM, which may impair the function of multiple respiratory chain complexes, including their ability to assemble in “supercomplexes.”28
Table 158-3. Syndromes, Genes, and Proteins Associated with mtDNA Depletion
 DEFECTS OF MITOCHONDRIAL DYNAMICS
DEFECTS OF MITOCHONDRIAL DYNAMICS
In most tissues, including the CNS, mitochondria are extremely dynamic organelles, which move constantly, split by fission, and fuse with one another: This gives mitochondria the ability to travel very long distances (eg, from the soma of a spinal cord motor neuron to the neuromuscular junction of a motor nerve in the toe), to spread energy in different areas of the cell, and to regulate their survival through mitophagy.29
Although the respiratory chain may be affected only indirectly, disorders of mitochondrial dynamics are being described with increased frequency and generally affect the optic nerve (dominant optic atrophy due to mutations in OPA1), the peripheral nerve (CMT type 2 due to mutations in MFN2), or the long tracts in the CNS (hereditary spastic paraplegia [HSP]). There is also good evidence that defects of mitochondrial motility may play a role in the pathogenesis of common neurodegenerative disorders,30 but this is beyond the scope of this chapter.
DIAGNOSTIC EVALUATION
The striking clinical heterogeneity of mitochondrial disorders poses a diagnostic challenge. The following five criteria may be of some help (Table 158-3).
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
As mentioned earlier, the complexity of the clinical presentation can direct attention to mutations in mtDNA, especially when certain telltale symptoms coexist: short stature, neurosensory hearing loss, progressive external ophthalmoplegia (PEO), axonal neuropathy, diabetes mellitus, hypertrophic cardiomyopathy, or renal tubular acidosis. On the other hand, it is equally important not to exclude mtDNA mutations in patients who have involvement of a single tissue, such as myopathy, cardiomyopathy, or renal disease. If the renal involvement affects glomerular rather than tubular function, the possibility of CoQ10 deficiency should be considered, as many CoQ10 deficiencies are associated with nephrosis.
 INHERITANCE
INHERITANCE
Because most mtDNA-related disorders are maternally inherited but clinical expression is extremely variable, it is crucially important to collect a meticulous family history, with special attention to soft signs in maternal relatives, such as short stature, migraines, deafness, and diabetes. However, lack of maternal inheritance does not exclude the diagnosis (such as in cases of Kearns-Sayre syndrome (KSS) and myopathy, discussed previously). In children with Leigh syndrome, inheritance is an important initial discriminator between mtDNA-related forms (MILS; mutations in ND5 or ND3) and Mendelian forms (complex I subunits, COX assembly factors, CoQ10 deficiency, defects of mtDNA translation).
 LABORATORY FINDINGS
LABORATORY FINDINGS
Lactic acidosis is a common finding in defects of the respiratory chain, and the lactate-to-pyruvate ratio is usually elevated (50:1 to 200:1, compared to a normal ratio of 25:1). Information about cerebrospinal fluid (CSF) lactate and pyruvate is important, because CSF values can be increased in children with encephalopathy and normal blood lactate. However, lack of lactic acidosis does not exclude the diagnosis. For example, in patients with NARP or maternally inherited Leigh syndrome (MILS), blood lactate and pyruvate can be normal or only mildly elevated. Serum creatine kinase (CK) levels are usually normal or modestly increased except, of course, in patients with myopathy during episodes of myoglobinuria. As another exception to this rule and as a useful diagnostic clue, serum CK values can be markedly elevated in children with the myopathic form of mtDNA depletion, such as those with mutations in the gene (TK2) encoding thymidine kinase. Another important laboratory test is serum thymidine, which is greatly increased in patients with MNGIE (see Table 158-3).
 NEURORADIOLOGY
NEURORADIOLOGY
Bilateral MRI signal hyperintensities in basal ganglia are typical of Leigh syndrome (LS). Strokelike lesions in the posterior cerebral hemispheres are typically seen in patients with MELAS. Diffuse signal abnormality of the central white matter is characteristic of KSS, whereas basal ganglia calcifications are common in both MELAS and KSS syndrome. Leukodystrophy, sometimes cavitating, has been associated with mutations in an assembly protein of complex I, with a newly described mutation in COX VIB1,16 and in defects of mtDNA translation. Proton magnetic resonance spectroscopy reveals lactate accumulation in the CSF and in specific areas of the brain, where lactate concentration can be compared to the concentration of N-acetyl-L-aspartate (NAA), an indicator of cell viability.
 EXERCISE PHYSIOLOGY
EXERCISE PHYSIOLOGY
Impaired oxygen extraction by exercising muscle can be detected by near-infrared spectroscopy, which measures the degree of deoxygenation of hemoglobin. A simpler test is based on measuring the partial pressure of oxygen (PO2) in cubital venous blood after forearm aerobic exercise. PO2 rises paradoxically in patients with mito-chondrial myopathy or PEO, and the degree of rise reflects the severity of oxidative impairment. By31 P-magnetic resonance spectroscopy, the ratio of phosphocreatine to inorganic phosphate (PCr to Pi) can be measured in muscle at rest, during exercise, and during recovery. In patients with mitochondrial dysfunction, PCr-to-Pi ratios are lower than normal at rest, they decrease excessively during exercise, and they return to baseline more slowly than normal.31
PATHOLOGY
Muscle biopsy plays a central role in diagnosing mitochondrial disorders. Abnormal mitochondrial proliferation, a hallmark of mitochondrial dysfunction, can be revealed with the modified Gomori trichrome stain (“ragged-red fibers” [RRF]) or, better, with the succinate dehydrogenase (SDH) reaction (“ragged-blue fibers”). RRF are present in muscle biopsies taken from patients with most disorders due to mtDNA mutations, in which—because of hetero-plasmy—they show a “checkerboard” pattern. Rare exceptions to this rule include NARP/MILS and Leber’s hereditary optic neuropathy. RRF are also present in Mendelian mitochondrial diseases, especially in those due to defects of intergenomic signaling.
The cytochrome c oxidase (COX) histochemical reaction typically reveals scattered COX-negative fibers, which usually correspond to—but are not confined to—RRF. RRF are COX-positive in two main conditions: (1) typical MELAS syndrome and (2) mutations in mtDNA genes encoding cytochrome b or complex I subunits. To confirm the pathogenicity of an mtDNA mutation, single fibers can be dissected out of thick cross-sections of muscle and used to determine the levels of a given mutation by polymerase chain reaction (PCR). Finding higher levels of the mutation in affected (ragged-red or COX-negative fibers) than in unaffected (non-ragged-red, COX-positive fibers) fibers is strong evidence that the mutation in question is pathogenic.
A simple and elegant stratagem to reveal COX-deficient (as opposed to COX-negative) fibers is to superimpose the SDH and COX stain: In normal conditions, the brown COX stain prevails in all fibers, whereas the blue SDH stain “shines through” in COX-deficient fibers. When the COX stain is uniformly decreased in all fibers (rather than in a mosaic pattern), this points toward nucleus-encoded defects of complex IV, such as SURF1 mutations in Leigh syndrome.
Electron microscopy has lost much of its historical diagnostic value, but it can reveal focal accumulations of mitochondria or mitochondrial with paracrystalline inclusions in cases in which histochemical results are equivocal.
The basic neuropathology in mitochondrial encephalomyopathies consists of four histological lesions: spongy degeneration, neuronal loss, gliosis, and demyelination. While these lesions are nonspecific, their distribution patterns vary in patients with different mtDNA-related disorders. Thus, the neuropathology of Kearns-Sayre syndrome is characterized by spongiform degeneration, which is usually generalized and affects both gray and white matter. In MELAS, multifocal necrosis predominates, affecting the cerebral cortex or the subcortical white matter, and the cerebellum, thalamus, and basal ganglia. Calcification of basal ganglia is also common. In MERRF, neuronal degeneration, astrocytosis, and demyelination affect preferentially the cerebellum (especially the dentate nucleus), the brain stem (especially the olivary nucleus), and the spinal cord. In LS, there are symmetrical, bilateral foci of necrosis in the basal ganglia, thalamus, midbrain, and pons. Microscopically, the lesions show vascular proliferation, gliosis, neuronal loss, demyelination, and cystic cavitation.
BIOCHEMISTRY
All 13 proteins encoded by mtDNA are components of the mitochondrial respiratory chain. Seven (ND1-ND5, ND4L, and ND6) are subunits of complex I, one (cytochrome b) is part of complex III, three (COX I, COX II, and COX III) are subunits of complex IV, and two (ATPase 6 and ATPase 8) are subunits of complex V (Fig. 158-2). Therefore, disorders caused by mutations of mtDNA (regardless of these being primary or secondary to defects of intergenomic communication) are usually associated with biochemical defects of oxidative phosphorylation. Skeletal muscle is the preferred tissue for biochemical analysis, because it is rich in mitochondria, it is invariably affected in multisystem disorders (mitochondrial encephalomyopathies), and it is the most common tissue affected in isolation (mitochondrial myopathies). There is some controversy about whether fresh muscle tissue is needed or if frozen tissue suffices. It is our view that—except for the rare cases in which freshly isolated mitochondria for polarographic analyses are required—frozen specimens provide adequate biochemical information. Because mitochondrial proliferation in muscle (ragged-red fibers) is a common occurrence, activities of respiratory chain complexes should be referred to the activity of citrate synthase, a nuclear-encoded enzyme of the mitochondrial matrix that is a good marker of mitochondrial abundance.
Biochemical studies in muscle extract from patients with mitochondrial disorders yield two main types of results. The first pattern consists of partial defects in the activities of multiple complexes containing mtDNA-encoded subunits (I, III, and IV), contrasting with normal or increased activities of SDH (complex II) and citrate synthase. The second pattern is a severe deficiency in the activity of one specific respiratory complex. As a rule, the first pattern is found in patients with single or multiple mtDNA deletions (eg, KSS, SANDO), mtDNA depletion (eg, Alpers syndrome), or mutations in tRNA genes (eg, MELAS, MERRF), because these genetic errors impair mitochondrial protein synthesis in toto. The second pattern is also typical of mutations in mtDNA protein-coding genes. For example, mutations in the cytochrome b gene cause isolated complex III deficiency, or mutations in SURF1 cause isolated COX deficiency. However, mutations in ND genes, which are encountered with increasing frequency in patients with MELAS, LS, Leber’s hereditary optic neuropathy, or overlap syndromes, cause inconsistent and often modest impairment of complex I activity.14
 MOLECULAR GENETICS
MOLECULAR GENETICS
Human mtDNA is a 16,569-bp circle of double-stranded DNA. It is highly compact and contains only 37 genes: 2 genes encode ribosomal RNAs (rRNAs), 22 encode transfer RNAs (tRNAs), and 13 encode polypeptides. Over 200 pathogenic mutations have been identified in mtDNA (Fig. 158-1).
However, a large—and steadily increasing—number of mutations have been found in nuclear DNA that affect subunits of the respiratory chain (RC) directly or indirectly (assembly proteins, mtDNA integrity, translation or replication, and MIM lipid milieu). Thus, the “menu” for the molecular definition of an RC disease has become very long, and it is impossible (and prohibitively expensive) to sample every item in the list. The first decision is seeing whether the mutation is more likely to be in mtDNA or in nDNA. Although MitoChips, which allow sequencing of mtDNA in its entirety, are becoming accessible, it is still more rational to decide—on the basis of the criteria outlined above—whether the mutation affects mtDNA protein synthesis in toto or if it affects individual mtDNA-encoded proteins. If the mutation is more likely to affect the nuclear DNA, clinical and biochemical data will help one decide which nuclear genes to sequence. For example, an infant who is the product of a consanguineous union and has a severe mitochondrial myopathy with multiple RC defects and markedly decreased content of mtDNA in muscle should be screened for a TK2 mutation. Accurate molecular diagnosis is of practical importance, because it offers the parents of children with lethal diseases the option of prenatal diagnosis for future pregnancies.
THERAPY
Therapy of mitochondrial diseases is still woefully inadequate.32 Besides palliative pharmacological and surgical interventions directed at alleviating symptoms, approaches to therapy include (1) removing toxic products, especially lactic acid; (2) administering artificial electron acceptors, such as vitamin K3 and vitamin C; (3) administering metabolites and cofactors, such as L-carnitine and coenzyme Q10 (CoQ10); and (4) administering oxygen radical scavengers, such as CoQ10. A simple and effective therapeutic approach is aerobic exercise, which prevents muscle deconditioning and improves functional and biochemical features of muscles harboring pathogenic mtDNA mutations.
Gene therapy is daunting in these conditions. However, if we could cause even a small shift in the relative proportion of mutant and normal mtDNAs, thus lowering the mutant load below the pathogenic threshold, we probably would dramatically improve the clinical expression. Various strategies are being considered, including using peptide nucleic acids (PNAs) to inhibit the replication of complementary mutant mtDNAs or using pharmacological approaches directed to eliminate mitochondria with high proportions of mutations. The observation that myoblasts often contain lesser amounts of pathogenic mtDNA mutations than mature muscle fibers has led to the use of myotoxic agents or isometric exercise to induce limited muscle damage, which is followed by regeneration of muscle fibers harboring lower mutational loads. Although these therapies have met with mixed results, they show our ability to affect mutational loads, at least in skeletal muscle.
Therapy of Mendelian mitochondrial disorders is also very limited. It is encouraging, however, that allogeneic bone marrow transplantation (the first stem cell therapy in this field) in a patient with MNGIE has resulted in steady clinical, laboratory, and biochemical improvement over 2 years.33
ACKNOWLEDGMENTS
Part of the work presented here was supported by National Institutes of Health grant PO1HD32062 and by the Marriott Mitochondrial Disorders Clinical Research Fund (MMDCRF).
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


