Fig. 62.1
Clinical photograph showing left-sided Wilms tumor
An abdominal swelling or mass
Abdominal pain
Fever
Nausea and vomiting
Hematuria
High blood pressure
The tumor can grow rapidly, which may result from bleeding into the tumor or from actual tumor growth.
Hematuria:
May be seen in 10–15 % of cases.
This is seen often after relatively minor trauma related to injury of an enlarged kidney involved by tumor.
Hematuria may be also a late presentation that is usually associated with tumor invasion of the calyces and is considered a bad prognostic sign.
Hypertension:
Increased blood pressure may be present in 20 % of cases.
Hypertension in Wilms’s tumor results from pressure effect of the tumor on the renal vessels leading to increased secretion of rennin.
Rarely, the tumor may produce erythropoietin leading to increased red blood cell production.
Wilms tumor can occur as part of rare genetic syndromes, including:
1.
Wilms tumor , aniridia , genitourinary anomalies, and mental retardation (WAGR) syndrome. This syndrome includes:
◦ Wilms tumor
◦ Aniridia
◦ Abnormalities of the genitals and urinary system
◦ Mental retardation
2.
Denys–Drash syndrome. This syndrome includes:
◦ Wilms tumor
◦ Kidney disease
◦ Male pseudohermaphroditism
◦ These patients mostly have bilateral or multiple tumors
3.
Beckwith–Wiedemann syndrome. This syndrome includes:
◦ Omphalocele
◦ A large tongue (macroglossia)
◦ Enlarged internal organs
Investigations
These include a complete blood count, blood urea nitrogen (BUN), creatinine, abdominal X-ray, abdominal ultrasound, computed tomography (CT) scan of the chest and abdomen, and magnetic resonance imaging (MRI; Figs. 62.2, 62.3, 62.4, and 62.5).
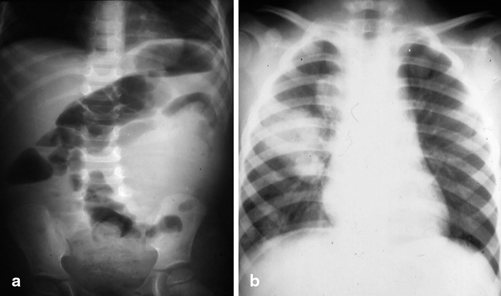
Fig. 62.2
Abdominal (a) and chest (b) X-rays in a patient with left-sided Wilms tumor. Note the soft tissue density pushing the bowel to the right and secondaries in the chest
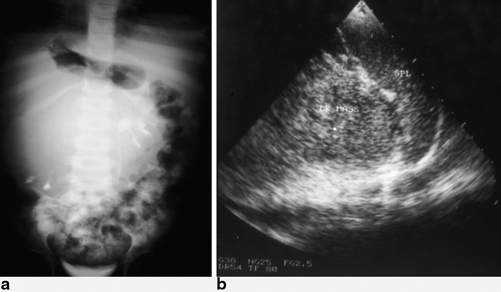
Fig. 62.3
An intravenous urography in a patient with right-sided Wilms tumor (a). Note the mass displacing and compressing the remaining calyces downward. Abdominal ultrasound in a patient with a left-sided Wilms tumor (b)
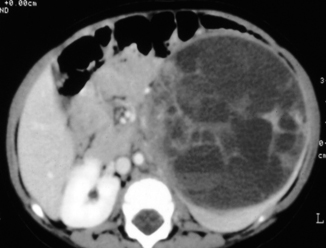
Fig. 62.4
Abdominal CT scan showing a large left-sided Wilms tumor. Note the thin rim of remaining kidney, which confirms that the tumor is arising from the kidney as opposed to neuroblastoma
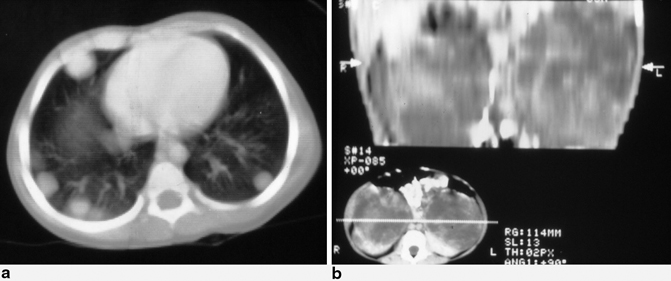
Fig. 62.5
Chest CT scan in a patient with Wilms tumor showing bilateral multiple secondaries (a) and abdominal CT scan showing bilateral Wilms tumor (b)
The aim is to evaluate the site, size, and extent of the tumor as well as the presence or absence of secondaries.
This as well as the function of the affected and contralateral kidney.
The presence or absence of synchronous tumors.
It is of great importance to exclude extension of the tumor into the renal vein as well as the inferior vena cava.
A plain abdominal radiograph often shows displacement of abdominal organs and occasionally the presence of calcification (< 10 %). The calcification usually is located on the edge of the tumor, whereas with a neuroblastoma, the calcification is speckled throughout.
Abdominal ultrasound confirms that the kidney is the site of the tumor, determines whether the mass is a cystic or solid tumor, and indicates if the tumor extends into the renal veins and inferior vena cava. Doppler ultrasound is a valuable investigation in detecting tumor extension in the renal vein and inferior vena cava.
CT scan defines the Wilms tumor site; identifies the presence of enlarged lymph nodes; evaluates the possible presence of a second Wilms tumor in the opposite kidney; assesses involvement of the tumor into the renal veins, inferior vena cava, and right atrium, and determines if the patient has intra-abdominal secondaries to the liver.
A chest CT is obtained to evaluate the presence of secondaries in the lungs.
Small abnormalities seen on chest X-ray are suggestive of secondaries, but those seen on CT scan may need to be confirmed by biopsy.
With the current radiological investigations, physical inspection of the opposite kidney by opening Gerota’s fascia as suggested previously to check for synchronous tumor is no longer necessary.
Pathology
Grossly, Wilms tumor is a large, solitary, and well-circumscribed mass with the remaining rim of normal kidney tissue. There is usually a well-defined membrane between them. On cut section, Wilms tumor is soft, homogenous, and tan-gray in color and may contain areas of hemorrhage and necrosis (Fig. 62.6).
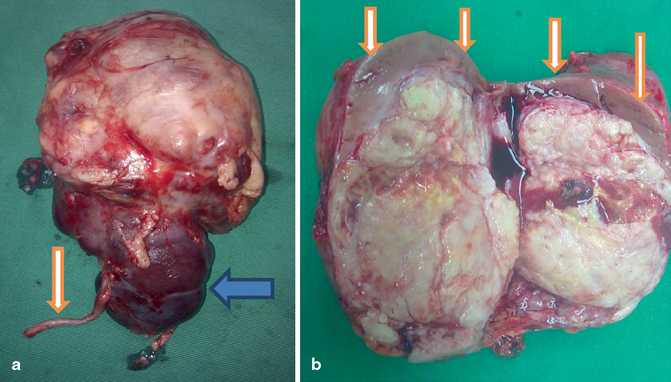
Fig. 62.6
a Clinical photograph showing a resected large Wilms tumor arising from upper pole. Note the ureter and lower pole. b Clinical photograph showing a bisected Wilms tumor. Note the small normal renal tissue at the upper pole of the kidney
Pathologically, Wilms tumor is a malignant tumor composed of three elements (a triphasic nephroblastoma) .
These include metanephric blastema, mesenchymal stroma, and epithelium.
Characteristic of Wilms tumor is the presence of abortive tubules and glomeruli surrounded by a spindled cell stroma. The stroma may include striated muscle, cartilage, bone, fat, or fibrous tissue.
The mesenchymal component may also include cells showing rhabdomyoid differentiation. The rhabdomyoid component may itself show features of malignancy (rhabdomyosarcomatous Wilms). This particular subtype shows poor response to chemotherapy.
Pathologically, Wilms tumors are divided into two prognostic groups:
1.
Favorable: Contains well-developed components.
2.
Unfavorable (Anaplastic): Contains anaplastic cells, which could be focal or diffuse. This is associated with higher frequencies of relapse and death.
Most cases of Wilms tumor do not have mutations in any of the genes.
A gene on the X chromosome, WTX, is inactivated in up to 30 % of Wilms tumor cases.
The gene WT1:
This is also called Wilms tumor-suppressor gene.
It has been found to make a protein that is found mostly in the fetal kidney and in tissues that give rise to the genitourinary system.
Inactivation of the gene may be responsible for the occurrence of Wilms tumor.
Mutations of the WT1 gene on chromosome 11 p 13 are observed in approximately 20 % of Wilms tumors.
At least half of the Wilms tumors with mutations in WT1 also carry mutations in CTNNB1, the gene encoding the proto-oncogene beta-catenin. Staging (National Wilms Tumor Study—NWTS). Post-surgical staging.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


