Renal Malformations
Norman D. Rosenblum
Renal malformations account for 30% to 50% of end-stage renal disease in children. Approximately one half of such cases are associated with lower urinary tract abnormalities. A spectrum of phenotypes exists and has been termed CAKUT (congenital abnormalities of the kidney and urinary tract). This chapter focuses on the epidemiology of these abnormalities and their etiologies, clinical manifestations, and aspects of clinical management.
CLASSIFICATION AND DEFINITION
Kidney malformation is classified at the clinical level by gross and microscopic anatomical features. One commonly used classification system is as follows:
• Renal agenesis—congenital absence of the kidney and ureter
• Simple renal hypoplasia—small kidney with a reduced number of nephrons and normal renal architecture
• Renal dysplasia—presence of malformed kidney tissue elements
• Renal dysplasia/hypoplasia (hypodysplasia)—small, dysplastic kidney
Characteristic microscopic features of the dys-plastic kidney (Fig. 469-1) include abnormal differentiation of mesenchymal and epithelial elements; a decreased number of nephrons; loss of corticomedullary differentiation; and the presence of dysplastic elements, including cartilage and bone. Dysplastic kidneys range in size from large distended kidneys with multiple large cysts to small kidneys, with or without cysts. A small dysplastic kidney without macroscopic cysts is often classified clinically as a hypoplastic/dysplastic kidney, since pathological examination, which provides a means to distinguish between simple hypoplasia and dysplasia, is not commonly performed during life. The multicystic dysplastic kidney (MCDK) is an extreme form of renal dysplasia.
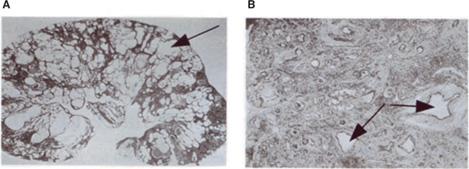
FIGURE 469-1. Microscopic features of human renal dysplasia. A: Multicystic dysplastic kidney characterized by numerous cysts (arrow) distorting the renal architecture. B: Dysplastic renal tissue demonstrating lack of recognizable nephron elements, dilated tubules, large amounts of stromal tissue, and primitive ducts (arrows) characterized by epithelial tubules with fibromuscular collars.
EPIDEMIOLOGY
The majority of renal malformations are detected during the antenatal period by fetal ultrasound and account for 20% to 30% of all anomalies detected. The incidence of renal and urinary tract malformations is 0.3 to 1.6 per 1000 liveborn and stillborn infants.1 Renal agenesis and dysplasia is 1.3- to 1.9-fold more common in males than in females.2 Fifty percent of individuals with a kidney malformation have a lower urinary tract anomaly. Of these, 25% have vesicoureteral reflux, 11% have ureteropelvic junction obstruction, and 11% are affected by ureterovesical junction obstruction.3 Renal malformations, other than mild antenatal pelviectasis, are associated with malformation of nonrenal tissues in about 30% of cases.1 There are over 100 syndromes associated with renal and urinary tract malformations. The most frequent syndromes are cited in Table 469-1. Bilateral renal agenesis occurs in 1 in 3000 to 10,000 births, while unilateral renal agenesis is estimated to have a prevalence of 1 in 1000. The incidence of unilateral dysplasia is 1 in 3000 to 5000 births (1:3640 for the MCDK) compared to 1 in 10,000 for bilateral dysplasia. Nine percent of first-degree relatives of patients with bilateral renal agenesis or bilateral renal dysgenesis have some type of renal malformation.4 The incidence of renal ectopia is 1 in 1000 autopsies, but the clinical recognition is estimated to be only 1 in 10,000 patients. Males and females are equally affected. Renal ectopia is bilateral in 10% of cases; when unilateral, there is a slight predilection for the left side. The incidence of fusion anomalies is estimated to be about 1 in 600 infants.
Table 469-1. Most Frequent Syndromes with Renal or Urinary Tract Malformation
Beckwith-Wiedemann |
Cerebro-oculo-renal |
CHARGE |
DiGeorge |
Ectrodactyly, ectodermal dysplasia and cleft/lip palates |
Ehlers-Danlos |
Fanconi pancytopenia syndrome |
Fraser |
Fryns |
Meckel |
Marfan |
MURCS Association |
Oculo-auriculo-vertebral (Goldenhar) |
Oculo-facial-digital (OFD) |
Pallister-Hall |
Renal cyst and diabetes |
Simpson-Golabi-Behmel (SGBS) |
Tuberous sclerosis |
Townes-Brocks |
Vertebrae, anus, trachea, espophagus, and renal (VATER) |
Wilms’ tumor, aniridia, genitourinary malformations, mental retardation (WAGR) |
Williams-Beuren |
Zellweger (cerebrohepatorenal) |
CHARGE, Coloboma, heart defect, retardation of growth, genital and ear anomalies; VATER, vertebral anomalies, anal atresia, tracheo-esophageal fistula, renal anomalies; WAGR, Wilms tumor, aniridia, genitourinary anomalies, retardation.
GENETIC CAUSES
Over 30 genes have been identified in children with syndromic forms of renal malformation (Table 469-2). For a complete list of syndromes featuring renal malformations, the reader is referred to McKusick’s Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/). Emerging evidence suggests that sporadic cases are associated with genetic mutations as well. Below, examples of genetic mutations associated with syndromic and nonsyndromic forms are discussed. The reader is also referred to Chapter 464, which covers the functions of these and other genes during renal development.
 BRANCHIO-OTO-RENAL (BOR) SYNDROME
BRANCHIO-OTO-RENAL (BOR) SYNDROME
BOR Syndrome, defined by the association of branchial (B), otic (O), and renal (R) anomalies, is an autosomal dominant disorder with incomplete penetrance and variable expressivity5(OMIM 113650). However, individual patients may have only one or two of these major features in association with other minor features such as external ear anomalies, preauricular tags, or other facial abnormalities (eg, BO syndrome, OMIM 120502). Renal malformations include unilateral or bilateral renal agenesis; hypodysplasia; and malformation of the lower urinary tract, including vesicoureteral reflux, pyeloureteral obstruction, and ureteral duplication. Different renal malformations can be observed in the same family. The genes EYA1 and SIX1 are mutated in BOR syndrome.6,7
Table 469-2. Human Gene Mutations Exhibiting Defects in Renal Morphogenesis
 RENAL-COLOBOMA SYNDROME (RCS)
RENAL-COLOBOMA SYNDROME (RCS)
Renal coloboma syndrome (RCS), an autosomal dominant disorder, is characterized by renal hypoplasia, vesicoureteric reflux, and optic nerve coloboma. A wide range of renal and ocular malformations is observed in RCS. Renal hypoplasia and vesicoureteric reflux are most frequent, but multicystic dysplasia and pelviureteric junction obstruction have also been described.9 RCS is caused by mutations in PAX2,10 which encodes a transcription factor that belongs to the paired-box family of homeotic genes. 
 RENAL CYST AND DIABETES SYNDROME (RCAD)
RENAL CYST AND DIABETES SYNDROME (RCAD)
RCAD is defined by the presence of renal hypodysplasia with cysts and diabetes mellitus. Occasional features include genital malformations. Diabetes mellitus, which is present in approximately 60% of all the cases reported, usually occurs before 25 years of age and is often associated with pancreatic atrophy.11 The presence of cysts is the most consistent feature of the renal pheno-type, leading to the name renal cysts and diabetes syndrome. The cysts are usually cortical, bilateral, and small.12 Mutations in TCF2 encoding the transcription factor HNF1b are present in affected patients.13 Mutations in TCF2 are being increasingly identified in patients with renal hypoplasia and dysplasia, multicystic dysplastic kidneys, renal agenesis, horseshoe kidneys, pelviureteric junction obstruction, and clubbing and tiny diverticula of the calyces. When histology is available, the most specific finding is the presence of cortical glomerular cysts with dilatation of the Bowman spaces (glomerulocystic dysplasia). Other nonspecific lesions such as cystic renal dysplasia, interstitial fibrosis, or oligomeganephronia have also been reported. Antenatal presentation with enlarged hyperechoic kidneys seems to be a quite common finding.14 More than 50 mutations have been reported in TCF2. There is no correlation between the type of mutation and the phenotype. Importantly, deletions frequently arise de novo in the proband. As observed in other syndromes, phenotypic variability can be observed between families and in family members with the same mutation.
 TOWNES-BROCK SYNDROME (TBS)
TOWNES-BROCK SYNDROME (TBS)
TBS is an autosomal dominant malformation syndrome characterized by imperforate anus; preaxial polydactyly; or triphalangeal thumbs, external ear defects, sensorineural hearing loss, and, less frequently, kidney, urogenital, and heart malformations.15,16 REAR syndrome (renal-ear-anal-radial) has also been a term used to describe this condition.17 The gene mutated in human TBS is SALL1, a member of the Spalt family that is required for the normal development of the limbs, nervous system, kidney, and heart.18
 RENAL TUBULAR DYSGENESIS (RTD)
RENAL TUBULAR DYSGENESIS (RTD)
RTD is a severe perinatal disorder characterized by the absence or paucity of differentiated proximal tubules, early severe oligohydramnios, and perinatal death usually due to pulmonary hypoplasia and skull ossification defects.19 It may be acquired during fetal development or inherited as an autosomal recessive disease. Autosomal recessive renal tubular dysgenesis is genetically heterogeneous and linked to mutations in the genes that encode components of the renin-angiotensin system.20 This condition also occurs in fetuses exposed in utero to angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (Ang II) receptor antagonists.
 GENETIC CAUSES OF ISOLATED (NONSYNDROMIC) RENAL MALFORMATION
GENETIC CAUSES OF ISOLATED (NONSYNDROMIC) RENAL MALFORMATION
In the majority of children with renal malformation, neither a syndrome nor a Mendelian pattern of inheritance is obvious. It has recently become apparent that mutations in genes that control renal development can be identified in many such patients. For example, analysis of a cohort of 100 patients with renal hypodysplasia and renal insufficiency demonstrated that 16% had mutations in one gene encoding for a transcription factor. The majority of mutations were identified in TCF2 (especially in the subset with kidney cysts) and PAX2. EYA1 and SALL1 mutations were found in single cases.8 Some of the mutations that were identified in these genes were de novo mutations, which explains the sporadic appearance of renal malformations. Careful analysis of patients with TCF2 and PAX2 mutations revealed the presence of extrarenal symptoms in only half; this supports previous reports that TCF2 and PAX2 mutations can be responsible for isolated renal tract anomalies or at least CAKUT malformations with minimal extrarenal features.12,21 This study demonstrates that subtle extrarenal symptoms in syndromal renal malformations can easily be missed.
Mutations in RET, which encodes a tyrosine kinase cell surface receptor, have been detected in patients with renal malformations. These mutations (Y791F and S649L) have also been detected in individuals with medullary thyroid carcinoma (MTC). None of the patients or their carrier relatives had clinical evidence of MTC at the time of the study. A low penetrance of renal malformation was suggested by minimal or no renal phenotypes in most carrier relatives.
MANAGEMENT
The widespread use and sensitivity of fetal ultrasound has increased the prenatal diagnosis of renal malformations. The sensitivity of prenatal ultrasound screening for renal malformations is about 82%, and the mean time at which these malformations are detected is 23 weeks gestation.1 In general, urinary tract malformations detected antenatally are isolated and present as mild hydronephrosis with no therapeutic consequences. Parents should be reassured. In contrast, bilateral forms of renal agenesis, severe dysgenesis, bilateral ureteric obstruction, or obstruction of the bladder outlet or the urethra can cause severe oligohydramnios as early as 18 weeks. Because amniotic fluid is critical to lung development, oligohydramnios as early as the second trimester can result in lung hypoplasia, a potentially fatal disorder. The oligohydramnios sequence, termed Potter’s syndrome, in its most severe form consists of a typical facial appearance characterized by pseudoepicanthus; recessed chin; posteriorly rotated, flattened ears and flattened nose; decreased fetal movement; clubfoot and clubhand; hip dislocation; joint contractures; and pulmonary hypoplasia. Severe renal malformations are usually the cause of oligohydramnios, which gives rise to the facial and limb deformities and impairs normal lung development. However, nonurinary causes such as amniotic fluid leakage and placental pathology could provoke oligohydramnios with the same consequences.
The renal prognosis can be evaluated antena-tally, with poor outcome likely when there is severe oligohydramnios and small, hyperechogenic kidneys. Antenatal diagnosis and assessment of the renal prognosis are important for considering early termination in cases of fatal or eventually severe renal disease and to prepare parents and medical staff for the likelihood of neonatal renal insufficiency. Other organ malformations should be sought carefully, and if detected, a karyotype should be done.
The clinical presentation of renal malformation in the postnatal period depends on the amount of functioning renal mass, the presence of bilateral urinary tract obstruction, and the occurrence of urinary tract infection. Bilateral renal agenesis or severe dysplasia is likely to present soon after birth with decreased renal function. This may be accompanied by oliguria. Alternatively, patients may present with a flank mass or an asymptomatic abnormality detected by renal imaging.
A detailed history and careful physical examination should be carried out on all infants with an antenatally detected renal malformation. An early renal ultrasound (within 24 hours of life) is recommended for newborns with a history of oligohydramnios, progressive antenatal hydronephrosis, distended bladder on antenatal sonograms, and bilateral severe hydroureteronephrosis. In male infants, a distended bladder and bilateral hydroureteronephrosis may be secondary to posterior urethral valves, a condition that requires immediate renal imaging and clinical intervention. In general, unilateral anomalies do not require urgent investigation after birth. Rather, renal ultrasound is recommended after the first 72 hours of life, because urine output gradually increases over the first 24 to 48 hours of life as renal plasma flow and glomerular filtration rate increase. Thus, the degree of urinary tract dilatation can be underestimated if performed during this period of transition.
Genetic testing in children with renal malformations should be preceded by a thorough clinical evaluation for extrarenal symptoms, including eye, ear, and metabolic anomalies. The presence of nonrenal anomalies increases the likelihood of detecting a specific genetic abnormality (Table 469-3). In addition, mutations in genes that are usually associated with syndromes can occur in patients with isolated renal malformations.
CLINICAL APPROACH TO SPECIFIC MALFORMATIONS
 UNILATERAL RENAL AGENESIS
UNILATERAL RENAL AGENESIS
A diagnosis of unilateral renal agenesis depends on the certainty that a second kidney does not exist in the pelvis or some other ectopic location. Since absence of one kidney induces compensatory hypertrophy in the existing kidney, the presence of a large kidney on one side suggests the possibility of unilateral renal agenesis. This condition is associated with contralateral urinary tract abnormalities, including ureteropelvic junction obstruction and vesicoureteral reflux in 20% to 40% of the cases. Thus, imaging of the contralateral side by ultrasonography and a voiding cystourethrogram is suggested. Managing affected patients involves determining the functional status of the contralateral kidney. A normal contralateral kidney is associated with an excellent long-term renal functional outcome. Since some studies have revealed that a substantial proportion of patients will develop proteinuria and hypertension in the long term, affected individuals should have their blood pressure measured and their urine periodically tested for protein throughout life.
Table 469-3. Clinical Indications to Search for a Renal Anomaly



