Pulmonary Alveolar Proteinosis
Bruce C. Trapnell and Lisa R. Young
Pulmonary alveolar proteinosis (PAP) is a syndrome characterized by the accumulation of surfactant lipids and proteins within the pulmonary alveoli, resulting in impaired gas exchange and respiratory insufficiency.1 Our understanding of PAP has advanced significantly over the past several decades due to a series of contributions from basic, clinical, and translational research.2,3 These studies have revealed a critical role for pulmonary granulocyte/macrophage-colony stimulating factor (GM-CSF) in the terminal differentiation of alveolar macrophages and in alveolar macrophage-mediated surfactant catabolism, pulmonary surfactant homeostasis, and lung host defense.4-6 PAP comprises part of a larger group of disorders associated with disruption of surfactant homeostasis and includes disorders of surfactant production (hereafter referred to as pulmonary surfactant metabolic dysfunction disorders) and disorders of surfactant clearance (hereafter referred to as PAP; Table 518-1). The distinct epidemiological, pathogenic, clinical, and prognostic features of these two disease categories indicate they are usually considered separately rather than as a continuum of a single disease process. PAP can be further divided into primary and secondary PAP, which, respectively, are associated with either loss of GM-CSF signaling or the presence of an underlying disorder that reduces alveolar macrophage numbers or functions.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Autoimmune pulmonary alveolar proteinosis (PAP) is the most common clinical form, accounting for 90% of all cases, and has an incidence and prevalence of 0.49 and 6.2 per million, respectively, in the general population.2,15 It typically presents in the third or fourth decade, and it is rarely observed in children under 10 years old.15,23 Secondary PAP is the next most common, accounting for 9% to 10% of cases, with an incidence and prevalence of 0.05 and 0.5 per million, respectively. Secondary PAP is linked to the occurrence of the underlying clinical conditions that cause it (Table 518-1). PAP caused by genetic mutations appears to comprise less than 1% of cases.
Pulmonary surfactant metabolic dysfunction disorders, which have varying degrees of surfactant accumulation, occur in individuals with mutations in the genes encoding surfactant protein B (SP-B), ATP-binding cassette A3 (ABCA3), and SP-C (SFTPB, ABCA3 and SFTPC genes, respectively). SP-B deficiency due to autosomal recessive SFTPB mutations is estimated to occur in 1 in 1.5 million births.11,25,26 ABCA3 dysfunction and pulmonary disease due to autosomal recessive ABCA3 mutations is predicted to occur more commonly than mutations causing SP-B deficiency.11,27 Population studies of SFTPC mutations have not been reported.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Surfactant is vital to lung structure and function and acts at the alveolar wall’s air-liquid-tissue interface to reduce surface tension, thereby preventing alveolar collapse and transudation of capillary fluid into the alveolar lumen. Surfactant lipids and proteins are synthesized, stored, and secreted into alveoli by alveolar type II epithelial cells. They are cleared by uptake and recycling in alveolar type II cells and by uptake and catabolism in alveolar macrophages.5 Surfactant pool size is tightly regulated by coordinate mechanisms controlling its synthesis, recycling, and catabolism.
Table 518-1. Classification of Disorders Associated with Disruption of Surfactant Homeostasis
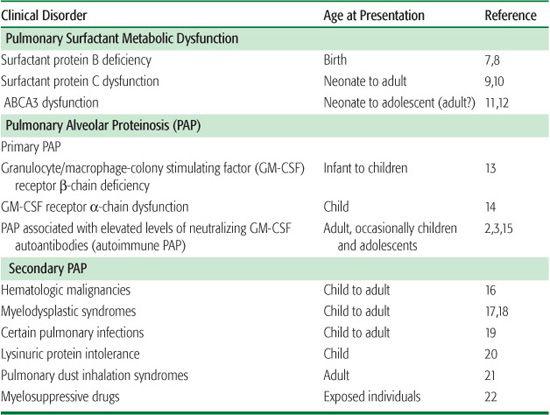
The pathogenesis of disorders of surfactant homeostasis is usefully considered in terms of defects in the production or clearance of pulmonary surfactant. PAP, an example of the latter, appears to result from reduced alveolar macrophage-mediated pulmonary clearance of surfactant. This can occur due to a reduction in the alveolar macrophage’s ability to catabolize surfactant lipids and proteins (ie, reduced intrinsic clearance) or to a reduction in their numbers (ie, reduced clearance capacity).
The first real clue about the pathogenesis of PAP was the observation that mice deficient in GM-CSF developed PAP due to a decrease in the alveolar macrophages’ ability to catabolize surfactant.6,36 Although GM-CSF deficiency has not been observed in humans,24 90% of PAP patients have high levels of neutralizing GM-CSF autoantibodies, which eliminate GM-CSF bioactivity in vivo and presumably mediate pathogenesis by blocking GM-CSF signaling to alveolar macrophages. The recent identification of familial PAP in association with recessive mutations of the GM-CSF receptor a chain supports the critical role of GM-CSF signaling in surfactant homeostasis and the pathogenesis of PAP in humans.
Several clinical disorders have been associated with the development of secondary PAP (Table 518-1). Although not well studied, the mechanism appears to be caused by either a reduction in the intrinsic ability of alveolar macrophages to catabolize surfactant or a reduction in the number of alveolar macrophages (resulting in a reduction in the capacity of the pulmonary alveolar macrophage population to catabolize surfactant). For example, myelofibrosis that impairs macrophage functions and chemotherapy sufficient to cause prolonged neutropenia can also reduce the numbers of otherwise intrinsically functional alveolar macrophages, resulting in decreased clearance capacity and surfactant accumulation.
Surfactant metabolic dysfunction disorders due to a deficiency or dysfunction of SP-B,41 SP-C,9 or ABCA311 result in production of abnormal surfactant, which alters lung mechanics and causes respiratory distress syndrome in infants and interstitial lung disease in older patients. The pathological features of these disorders include fibrosis, alveolar wall thickening, and significant parenchymal lung distortion (Fig. 518-1). This differs markedly from the pathological features of primary PAP, which in most cases is comprised primarily of the alveoli filling with surfactant without significant alveolar wall pathology (Fig. 518-1).
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
The clinical presentation in disorders of surfactant homeostasis differs widely, providing important initial clues to the underlying etiology. In primary pulmonary alveolar proteinosis (PAP), symptoms typically occur when surfactant has accumulated to sufficient levels to impair pulmonary gas exchange, at which time dyspnea develops insidiously. Autoimmune PAP typically presents as dyspnea in a previously healthy adult who may or may not have a history of smoking or other pulmonary exposures. Cough and production of whitish sputum are less common, and fever, hemoptysis, and chest pain are unusual unless infection is also present. Clubbing is uncommon. In the few individuals with primary PAP due to GM-CSF receptor abnormalities, insidious onset of dyspnea occurs in infancy or childhood. Secondary PAP occurs in individuals with a prior illness usually known to be associated with development of PAP. Thus, a high degree of suspicion is helpful.
Pulmonary surfactant metabolic dysfunction disorders have a wide range of presentations. SP-B deficiency presents in term infants as severe unexplained respiratory distress requiring emergent intubation and mechanical ventilation. ABCA-3 deficiency may present similarly to SP-B deficiency with neonatal respiratory failure in a term infant, but it may also present subacutely as ILD with symptoms of tachypnea, dyspnea, cough, hypoxemia, and diffuse infiltrates in older children.11,12,30 The manifestations of SP-C mutations are highly variable and can present at birth, throughout childhood, or in adulthood. Clinical or histological evidence of proteinosis has not been reported in adult cases.9–11,28,30
The differential diagnosis of respiratory failure in a term neonate also includes developmental lung anomalies, including alveolar capillary dysplasia with misalignment of the pulmonary veins (ACD-MPV), pulmonary hypoplasia, pulmonary interstitial glycogenosis, and lymphatic or vascular disorders.30 The differential diagnosis of PAP in older children includes mutations in SP-C, ABCA-3, CSF2RA, CSF2RB; autoimmune PAP; secondary PAP; and other interstitial lung diseases without alveolar proteinosis. The differential diagnosis of PAP in adolescents and adults includes autoimmune PAP, secondary PAP, and mutations in SP-C and possibly ABCA3.
 DIAGNOSIS
DIAGNOSIS
The diagnostic evaluation of disorders of surfactant homeostasis depends on the clinical context (ie, whether the disease is congenital or acquired, stable or rapidly progressive, or has occurred in other family members). Assessment may include a history and a physical, a chest radiograph, computed tomography of the chest, arterial blood gas measurements, bronchoscopy with bronchoalveolar lavage and transbronchial biopsy, surgical lung biopsy, serum GM-CSF autoantibody testing, and genetic testing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


