Principles of Inborn Errors of Metabolism
Jean-Marie Saudubray and Joe T. R. Clarke
Over 400 human diseases that are due to inborn errors of metabolism are now recognized, and this number is constantly increasing. However, the incidence of inborn errors may well be underestimated, because diagnostic errors are frequent. Despite the relative abundance of new case reports, there is considerable evidence that many of these disorders remain undetected or misdiagnosed. Several factors conspire to make the clinical diagnosis of inborn errors of metabolism (IEM) difficult.
IEM are individually rare but collectively numerous. The recent application of tandem mass spectrometry (tandem MS) to newborn screening and prenatal diagnosis has enabled presymptomatic diagnosis for some IEM. However, for most IEM, neonatal screening tests are either too slow, too expensive, or too unreliable; consequently, a simple method of clinical screening is mandatory before initiating sophisticated biochemical investigations. The clinical diagnosis of IEM relies upon a limited number of principles:
• Consider IEM in parallel with other more common conditions; for example, sepsis or anoxic-ischemic encephalopathy in neonates, and intoxication, encephalitis, and brain tumors in older patients
• Be aware of symptoms that persist and remain unexplained after the initial treatment and the usual investigations have been performed
• Collect blood and urine samples at the right time in relation to an acute illness
• Suspect that any neonatal death may be due to an IEM, particularly deaths that are attributed to sepsis
• Carefully review all autopsy findings
• Do not confuse a symptom (such as peripheral neuropathy, retinitis pigmentosa, cardiomyopathy, etc) or a syndrome (such as Reye syndrome, Leigh syndrome, sudden infant death, etc) with etiology
• Remember that an IEM can present at any age, from fetal life to old age
• Know that although most genetic metabolic errors are hereditary and transmitted as recessive disorders, the majority of individual cases appear sporadically
• Initially consider inborn errors that are amenable to treatment (mainly those that cause intoxication). Do not miss a treatable disorder.
• In acute emergency situations, undertake first those few investigations that are able to diagnose treatable IEM: First take care of the patient (emergency treatment) and then the family (genetic counseling).
In this section, inborn errors amenable to treatment are printed in bold. Additional information and diagnostic checklists are available online.
 CLASSIFICATION
CLASSIFICATION
The vast majority of IEM involve abnormalities in enzymes and transport proteins. However, all the metabolic disorders can be divided into the following two large clinical categories.
Category 1 Includes disorders that either involve only one functional system (such as the endocrine system, immune system, coagulation factors, or lipoproteins) or affect only one organ or anatomic system (such as the intestine, renal tubules, erythrocytes, or connective tissue). Presenting symptoms are uniform (e.g., a bleeding tendency in coagulation factor defects or hemolytic anemia in defects of glycolysis), and the correct diagnosis is usually easy to guess even when the basic biochemical lesion gives rise to systemic consequences.
Category 2 Includes diseases in which the basic biochemical lesion either affects one metabolic pathway common to a large number of cells or organs (e.g., storage diseases due to lysosomal disorders, energy deficiency in mitochondrial disorders) or is restricted to one organ but gives rise to humoral and systemic consequences (e.g., hyperammonemia in urea cycle defects, hypoglycemia in hepatic glycogenosis). The diseases in this category have a great diversity of presenting symptoms. The specific disorders mentioned are discussed later in this section.
From a pathophysiological perspective, metabolic disorders from Category 2 can be divided into three diagnostically useful groups.
Group 1 Disorders that cause intoxication. This group includes inborn errors of intermediary metabolism that lead to acute or progressive intoxication from the accumulation of toxic compounds proximal to the metabolic block. In this group are inborn errors of amino acid catabolism (phenylketonuria, maple syrup urine disease, homocystinuria, tyrosinemia, etc), most organic acidurias (methylmalonic, propionic, isovaleric, etc), congenital urea cycle defects, sugar intolerances (galactosemia, hereditary fructose intolerance), metal intoxication (Wilson, Menkes, hemochromatosis, and porphyrias). All the conditions in this group share clinical similarities: They do not interfere with embryo and fetal development, and they present with a symptom-free interval and clinical signs of “intoxication,” which may be acute (vomiting, coma, liver failure, thromboembolic complications, etc) or chronic (failure to thrive, developmental delay, ectopia lentis, cardiomyopathy, etc).
Conditions that can provoke acute metabolic attacks include catabolism, fever, intercurrent illness, and ingestion of specific foods. Clinical expression is often both late in onset and intermittent. The diagnosis is straightforward and most commonly relies on plasma and urine amino acid, organic acid, and acylcarnitine chromatography. Most of these disorders are treatable and require the emergency removal of the toxin by special diets, extracorporeal procedures, or “cleansing” drugs (carnitine, sodium benzoate, penicillamine, etc).
Although the pathophysiology is somewhat different, the inborn errors of neurotransmitter synthesis and catabolism (monoamines, GABA, and glycine) and the inborn errors of amino acid synthesis (serine, glutamine, and proline/ornithine) can also be included in this group, because they share many characteristics: They are inborn errors of intermediary metabolism; their diagnosis relies on plasma, urine, and CSF investigations (amino acid, organic acid analyses, etc); and some are amenable to treatment even when the disorder is present in utero—for example, 3-phosphoglycerate dehydrogenase deficiency.1
Group 2 Disorders involving energy metabolism. These consist of inborn errors of intermediary metabolism with symptoms due, at least partly, to a deficiency in energy production or utilization within the liver, myocardium, muscle, brain, or other tissues. This group can be divided into mitochondrial and cytoplasmic energy defects. Mitochondrial defects are the most severe and are generally untreatable. They include the congenital lactic acidemias (defects of pyruvate transporter, pyruvate carboxylase, pyruvate dehydrogenase, and the Krebs cycle), mitochondrial respiratory-chain disorders, and the fatty acid oxidation (FAO) and ketone body defects. Only the latter are partly treatable. Cytoplasmic energy defects are generally less severe. They include disorders of glycolysis, glycogen metabolism and gluconeogenesis, hyperinsulinism, and glucose transporter defects (all treatable disorders); the more recently described disorders of creatine metabolism (partly treatable); and the new inborn errors of the pentose phosphate pathways (untreatable). Common symptoms in this group include hypoglycemia, hyperlactatemia, hepatomegaly, severe generalized hypotonia, myopathy, cardiomyopathy, failure to thrive, cardiac failure, circulatory collapse, sudden unexpected death in infancy, and brain involvement. Some of the mitochondrial disorders and pentose phosphate pathway defects can interfere with embryo and fetal development and can cause dysmorphism, dysplasia, and malformations. Diagnosis is difficult and relies on function tests, enzymatic analyses requiring biopsies or cell culture, and molecular analyses.
Group 3 Disorders involving complex molecules. This group includes cellular organelles and diseases that disturb the synthesis or the catabolism of complex molecules. Symptoms are permanent, progressive, independent of intercurrent events, and unrelated to food intake. This group includes lysosomal storage disorders, peroxisomal disorders, disorders of intracellular trafficking and processing such as alpha1-antitrypsin deficiency, carbohydrate-deficient glycoprotein (CDG) syndrome, and inborn errors of cholesterol and bile acid synthesis. Almost none of these are treatable acutely; however, enzyme replacement therapy is now available for several lysosomal disorders. Intracellular localization of all these disorders is presented in Figure 134-1.
 GENETICS
GENETICS
Although most of these disorders are inherited as autosomal recessive conditions, a significant minority are transmitted as X-linked recessive disorders and a few as dominant disease (Table 134-1). Many cases appear to be sporadic, given the small size of sibships in developed countries. Consanguinity is rare, and many affected patients are compound heterozygotes. In this case, the phenotype is generally driven by the less severe mutation. Mutations in the mitochondrial genome, which is inherited maternally, still make up a rapidly growing subgroup of IEM; these disorders exhibit unique genetics and clinical characteristics. Compared to many other genetic diseases in which the gene product is unknown or not fully recognized, almost all IEM were primarily identified because of a suggestive biochemical profile, confirmed by an enzymatic defect. These biochemical methods remain the basis of diagnostic procedures and assessment of management. Antenatal diagnosis is available for most of these conditions.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
A few metabolic disorders are recognized by newborn screening of the general population (as for phenylketonuria) or of at-risk families. Apart from these, there are four groups of clinical circumstances in which a metabolic disorder is possible:
1. Acute or chronic antenatal and neonatal period.
2. Later-onset acute and recurrent attacks of symptoms such as coma, ataxia, vomiting, and acidosis.
3. Chronic and progressive generalized symptoms that can be mainly gastrointestinal (chronic vomiting), failure to thrive, hypotonia, recurrent infections, muscular, or neurological (myopathy, developmental delay, neurological deterioration, epilepsy).
4. Specific and permanent presentations that can involve any organ or system (heart, liver, intestine, kidney, lungs, endocrine, immune and hematological system, bone, and collagen). Many IEM can present with a specific isolated symptoms, such as cardiomyopathy, hepatomegaly, lens dislocation, renal tubulopathy, hyperkeratotic plaques, and so on.
The first three groups are presented in the following sections. The specific presentations in the fourth group are available online (see eTable 134.1  ). All treatable disorders are printed in bold type.
). All treatable disorders are printed in bold type.
Neonatal and Early Infancy Period
 Acute Encephalopathy and Metabolic Crash
Acute Encephalopathy and Metabolic Crash
The neonate has a limited repertoire of responses to severe illness. IEM may present with nonspecific symptoms such as respiratory distress, hypotonia, poor sucking reflex, vomiting, lethargy, seizures—problems that can easily be attributed to sepsis or some other common cause. Death of a sibling previously from a similar IEM may have been attributed to sepsis, cardiac failure or intraventricular hemorrhage.
Group 1 disorders are illustrated by an infant born full term, who after a normal pregnancy and delivery and an initial symptom-free period, relentlessly deteriorates for no apparent reason and does not respond to symptomatic therapy. The interval between birth and clinical symptoms may range from hours to weeks. Investigations, routinely performed in sick neonates, include a chest x-ray, cerebrospinal fluid examination, bacteriological studies, and cerebral ultrasound examination; all yield normal results. This unexpected and “mysterious” deterioration after a normal initial period is the most important indication for this group of IEM. Careful reevaluation of the child’s condition is then warranted.
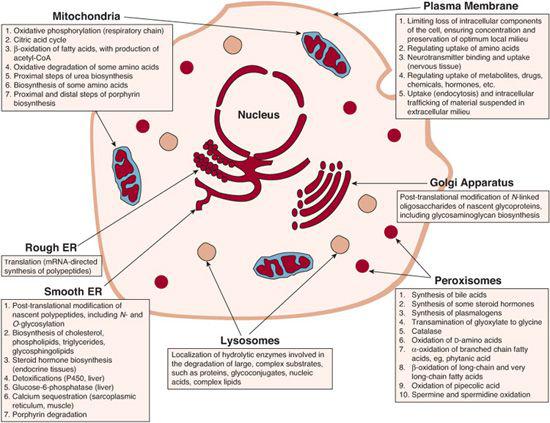
FIGURE 134-1. Cartoon showing intracellular localization of inborn errors of metabolism.
In Group 2 disorders (energy deficiencies), clinical presentation is often less striking and displays variable severity. A clinical algorithm for screening for treatable IEM in neonates is presented in Figure 134-2.
 Neurological Deterioration (Coma, Lethargy)
Neurological Deterioration (Coma, Lethargy)
This is the most frequent presenting sign in “intoxication” disorders. Typically, the first reported sign is poor sucking and feeding, after which the child sinks into an unexplained coma despite supportive measures. At a more advanced state, neurovegetative problems with respiratory abnormalities, hiccups, apneas, bradycardia, and hypothermia can appear. In the comatose state, characteristic changes in muscle tone and involuntary movements appear. Generalized hypertonic episodes with opisthotonus, boxing, or pedaling movements and slow limb elevations are observed in maple syrup urine disease (MSUD). As most nonmetabolic causes of coma are associated with hypotonia, the presence of “normal” peripheral muscle tone in a comatose child reflects a relative hypertonia. In organic acidurias, axial hypotonia and limb hypertonia with fast, large-amplitude tremors and myoclonic jerks (often mistaken for convulsions) are usual. An abnormal urine and body odor is present in some diseases in which volatile metabolites accumulate; for example, a maple syrup odor in MSUD and the sweaty-feet odor in isovaleric acidemia (IVA) and glutaric acidemia type II.
In energy deficiencies, the clinical presentation is less obvious and displays a more variable severity. In many conditions, there is no symptom-free interval. The most frequent findings are a severe generalized hypotonia; rapidly progressive neurological deterioration; and possible dysmorphism, or malformations. In contrast to the intoxication group, lethargy and coma are rarely initial signs. Hyperlactatemia with or without metabolic acidosis is frequent. Cardiac and hepatic involvement are also commonly associated with energy deficiencies.
In the neonatal period, only a few lysosomal storage disorders present with neurological deterioration. By contrast, most peroxisomal biogenesis defects present at birth with dys-morphism and severe neurological dysfunction (Chapter 162).
 Seizures
Seizures
Five treatable disorders can present in the neonatal period predominantly with intractable seizures: pyridoxine-responsive seizures, pyridox(am)ine-5′-phosphate oxidase deficiency, folinic acid–responsive epilepsy, 3-phosphoglycerate dehydrogenase deficiency responsive to serine supplementation, and persistent hyperinsulinemic hypoglycemia. Also, biotin-responsive holocarboxylase synthetase deficiency may rarely present predominantly with neonatal seizures. GLUT1 deficiency (brain glucose transporter), which is responsive to a hyperketotic diet, and biotin-responsive biotinidase deficiency can also present in the first months of life as epileptic encephalopathy.
Table 134-1. X-Linked Recessive and Dominant Inborn Errors of Metabolism (IEM)
(Treatable disorders shown in bold type) |
X-linked Recessive |
Glycogenosis type IX (phosphorylase b kinase) |
Pyruvate dehydrogenase E1-alpha subunit |
Cerebral creatine transporter (SLC6A8: creatine deficiency syndrome) |
Ornithine transcarbamylase (OTC: congenital hyperammonemia) |
X-linked dominant chondrodysplasia (cholesterol synthesis defect) |
CHILD syndrome (cholesterol synthesis defect) |
PRPP synthetase (de novo purine synthesis) |
Lesch-Nyhan disease (HGPRT: salvage purine pathway) |
X-linked sideroblastic anemia (porphyria) |
Menkes disease (copper transport) |
Fabry disease (alpha-galactosidase: sphingolipidosis) |
Hunter disease (mucopolysaccharidosis type II) |
X ALD/AMN (peroxisomal adrenoleukodystrophy) |
Allan-Herndon-Dudley syndrome (monocarboxylate acid tranporter 8: triiodothyronine transport) |
Dominant |
Most of the porphyrias but Gunther disease and hepatoerythropoietic porphyria |
Familial hypercholesterolemia (LDL receptor defect) |
Glutamic dehydrogenase hyperactivity (syndrome HI/HH: hyperinsulinism) |
Glucokinase hyperactivity (dominant hyperinsulinism) |
Renal glucosuria (SGLT2 glucose transporter) |
GLUT1 deficiency (cerebral glucose transporter) |
Hawkinsinuria (tyrosine catabolism) |
3-methyl crotonyl-glycinuria (leucine catabolism) |
GTP cyclohydrolase (biopterin synthesis defects) |
Many other untreatable inherited disorders can present in the neonatal period with severe epilepsy: nonketotic hyperglycinemia, D-glyc-eric aciduria, and mitochondrial glutamate transporter defect (all three presenting with myoclonic epilepsy and a burst-suppression EEG pattern), peroxisomal biogenesis defects, respiratory-chain disorders, sulfite oxidase deficiency, and Menkes disease. In all these conditions, epilepsy is severe; has an early onset; and can present with spasms, myoclonus, and partial or generalized tonic/clonic crises.
 Hypotonia
Hypotonia
Severe hypotonia is a common symptom in sick neonates. It is more generally observed in non-metabolic severe fetal neuromuscular disorders (Chapter 569). Only a few IEM present with isolated hypotonia in the neonatal period, and very few are treatable. The most severe metabolic hypotonias are observed in hereditary hyperlactatemia, respiratory-chain disorders, urea cycle defects, NKH, sulphite oxidase (SO) deficiency, peroxisomal disorders, and tri-functional enzyme deficiency. Severe forms of Pompe disease (alpha-glucosidase deficiency) can initially mimic respiratory-chain disorders or trifunctional enzyme deficiency when generalized hypotonia is associated with cardiomyopathy. However, Pompe disease does not strictly start in the neonatal period. Prader-Willi syndrome, one of the most frequent causes of isolated neonatal hypotonia at birth, can mimic hypotonia-cystinuria syndrome.5 These three neurological presentations are summarized in Table 134-2.
 Hepatic Presentation
Hepatic Presentation
Five main clinical groups of hepatic symptoms can be identified:
1. Hepatomegaly with hypoglycemia and seizures without liver failure suggest glycogenosis type I or III, gluconeogenesis defects, or severe hyperinsulinism.
2. Liver failure (jaundice, coagulopathy, hepato-cellular necrosis with elevated serum transaminases, and hypoglycemia with ascites and edema) suggests fructosemia (now rare because infant formulas are fructose free); galactosemia; tyrosinemia type I (after 3 weeks); neonatal hemochromatosis; respiratory-chain disorders; and transaldolase deficiency, a disorder of the pentose phosphate pathway that can present with hydrops fetalis. Severe fetal growth retardation, lactic acidosis, failure to thrive, hyperaminoaciduria, very high serum ferritin concentrations, hemosiderosis of the liver, and early death suggest GRACILE syndrome (Finnish lethal neonatal metabolic syndrome).6
3. Cholestatic jaundice with failure to thrive is a predominant finding in alpha1-antitrypsin deficiency, Byler disease, inborn errors of bile acid metabolism, peroxisomal disorders, Niemann-Pick type C disease, CDG (congenital disorder of glycosylation) syndromes, and citrin deficiency.7,8Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency can present early in infancy as cholestatic jaundice, liver failure, and hepatic fibrosis.
4. Hepatic presentations of inherited FAO (fatty acid oxidation) disorders and urea cycle defects consist of acute steatosis or Reye syndrome with normal bilirubin concentrations, slightly prolonged prothrombin time, and moderate elevation of transaminases rather than true liver failure. Investigating patients with severe hepatic failure is difficult with many pitfalls. At an advanced state, nonspecific abnormalities secondary to liver damage can be present. Mellituria (galactosuria, glycosuria, fructosuria), hyper-ammonemia, hyperlactatemia, hypoglycemia after a short fast, hypertyrosinemia (>200 μmol/L), and hypermethioninemia (sometimes higher than 500 μmol/L) are encountered in all cases of advanced hepato-cellular disease.
5. Hepatosplenomegaly (HSM) with other signs of storage disorders (coarse facies, macroglossia, hydrops fetalis, ascites, edema, dysostosis multiplex, vacuolated lymphocytes) are observed in lysosomal diseases. HSM with inflammatory syndrome, including hemato-logical or immunologic features, may be observed in lysinuric protein intolerance (macrophage-activating syndrome), mevalonic aciduria and leucopenia, (inflammatory syndrome and recurrent severe anemia), and transaldolase deficiency (hydrops fetalis with severe anemia).
 Cardiac Presentation
Cardiac Presentation
Some metabolic disorders can present predominantly with cardiac disease. Heart failure and a dilated hypertrophic cardiomyopathy, most often associated with hypotonia, muscle weakness, and failure to thrive, suggests fatty acid oxidation disorders, respiratory-chain disorders, or Pompe disease. Don’t miss a carnitine-uptake defect (systemic carnitine defect), which responds dramatically to carnitine administration. Some respiratory-chain disorders are tissue specific and are expressed only in the myocardium. Congenital disorders of glycosylation (CDG syndrome type Ia) can sometimes present in infancy with cardiac failure due to pericardial effusion, cardiac tamponade, and cardiomyopathy. Many defects of long-chain fatty acid oxidation can present with cardiomyopathy, arrhythmias, or conduction defects (atrioventricular block, bundle branch block, ventricular tachycardia), which may lead to cardiac arrest.9,10
 Initial Approach to Investigation
Initial Approach to Investigation
If clinical assessment suggests IEM, general supportive measures and laboratory investigations should be undertaken concurrently (see Table 134-3). Abnormal urine odor can be diagnostic.
Acetonuria (3+) in a newborn is always abnormal and is an important sign of a metabolic disease. Hypocalcemia and elevated or reduced blood glucose concentrations are frequently present in metabolic diseases. The physician should be wary of attributing marked neurological dysfunction purely to these findings.
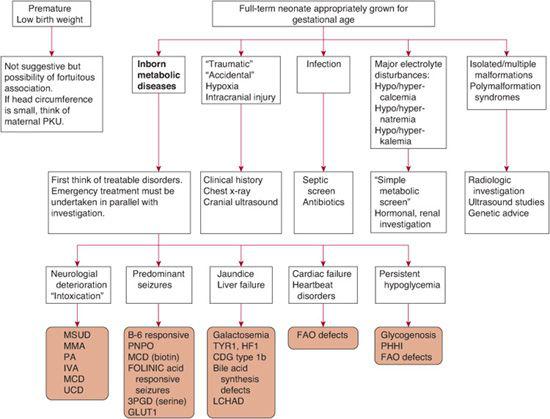
FIGURE 134-2. The “sick” neonate: An algorithm for screening for treatable inborn errors of metabolism. CDG, congenital disorders of glycosylation; FAO, fatty acid oxidation disorders; HFI, hereditary fructose intolerance; IVA, isovaleric acidemia; LCHAD, 3-hydroxy long chain acyl-CoA dehydrogenase; MCD, multiple carboxylase deficiency; MMA, methylmalonic aciduria; MSUD, maple syrup urine disease; PA, propionic acidemia; PHHI, primary hyperinsulinemic hypoglycemia of infancy; PKU, phenylketonuria; UCD, urea cycle defects; PNPO, pyridox(am)ine-5′-phosphate oxidase; 3PGD, 3-phosphoglycerate dehydrogenase; TYR-1, Tyrosinemia type 1.
The metabolic acidosis of organic acidurias is usually accompanied by an elevated anion gap. Urine pH should be below 5; otherwise, renal acidosis is a possibility. A normal blood pH does not exclude a moderate hyperlactatemia, which is significant in the absence of infection or tissue hypoxia. Blood ammonia and lactic acid concentrations should be determined systematically in at-risk newborns. Hyperammonemia with ketoacidosis suggests an underlying organic acidemia (OA). However, isolated hyperammonemia can occur, and an elevated ammonia level alone can induce respiratory alkalosis. Moderately elevated lactate concentrations (3–6 mmol/L) are often observed in organic acidemias and in the hyperammonaemias; levels greater than 10 mmol/L are frequent in hypoxia. With hypoxic lactic acidosis, the lactate to pyruvate ratio is < 20, and ketosis is absent. Propionic, methylmalonic, and isovaleric acidemias frequently present with granulocytopenia and thrombocytopenia, which may be mistaken for sepsis. Transaldolase deficiency and early onset forms of mevalonate kinase deficiency present with severe recurrent hemolytic anemia. Adequate amounts of plasma, urine, blood on filter paper, and cerebrospinal fluid (CSF) should always be stored, because they may later be important in establishing a diagnosis. Utilizing these precious samples should be carefully planned after advice from specialists in IEM.
After obtaining clinical and laboratory data, a process that should be completed within 2 to 4 hours, specific therapeutic recommendations can be made; this avoids long delays associated with waiting for the results of sophisticated diagnostic investigations. On the basis of this evaluation, most patients can be classified into one of five types (see Table 134-4). Some very significant symptoms (eg, metabolic acidosis and especially ketosis) can be moderate and transient, largely depending on the symptomatic therapy. Conversely, at an advanced state, many nonspecific abnormalities (eg, respiratory acidosis, severe hyperlactatemia, secondary hyperammonemia) can disturb the original metabolic profile. This applies particularly to IEM with a rapid fatal course, such as urea cycle disorders, in which the initial characteristic presentation of hyper-ammonemia with respiratory alkalosis shifts rapidly to a rather nonspecific picture of acidosis and hyperlactatemia.
We have found that more than 80% of newborns with inborn errors of intermediary metabolism have MSUD (maple syrup urine disease), organic acidurias, urea cycle defects, nonketotic hyperglycinemia, respiratory-chain, or fatty acid oxidation disorders.
 Approach to Therapy
Approach to Therapy
Immediate therapy of acute encephalopathy due to any of the likely IEM involves measures to decrease the production of offending metabolites and to increase their excretion. Treatment should include the following:
1. Ensure adequate cardiorespiratory function to allow removal of any accumulating metabolites. Adequate hydration is essential to maintain good urine output, because many of the offending diffusible metabolites are freely filtered at the glomerulus.
Table 134-2. Neurological Presentations in Neonate and in Early Infancy
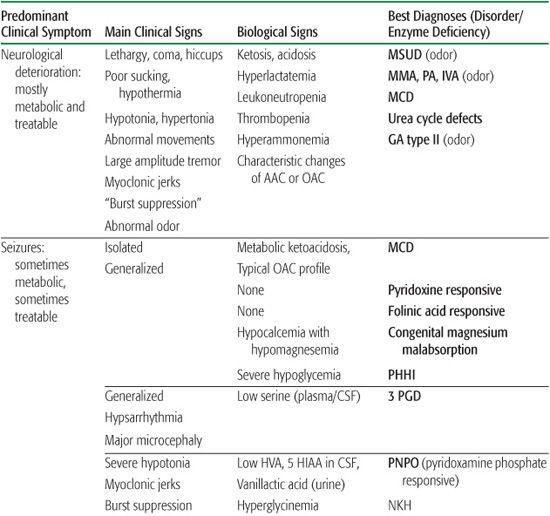
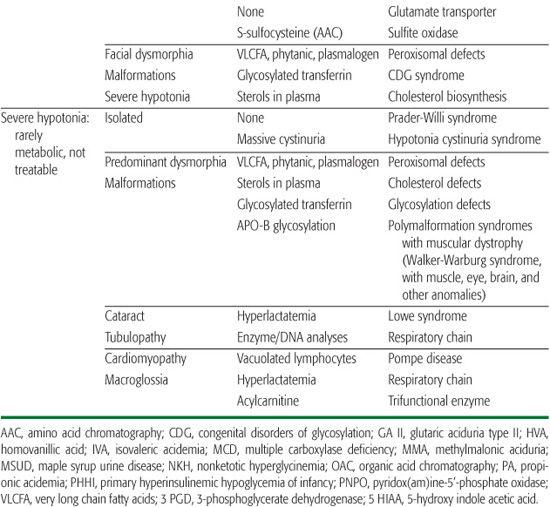
2. Reverse the catabolic state and reduce exposure to the offending nutrients. To achieve these first two therapeutic objectives, use 10% dextrose in 0.45% saline, with 20 mEq/L of potassium (if patient is voiding); run intravenously at 150% of maintenance fluid requirements. This regimen provides approximately 9 to 10 mg/kg per minute of glucose to neonates and infants. Fluid restriction may be necessary if cerebral edema is present.
3. Correct the metabolic acidosis by administering sodium bicarbonate if the serum bicarbonate level is less than 15 mEq/L. Beware of overcorrection: stop once the bicarbonate level reaches 15 mEq/L. Also, beware of iatrogenic hypernatremia, although this may be unavoidable.
4. After these measures have been instituted, and even before a precise biochemical diagnosis has been made, begin hemodialysis or hemofiltration to remove the offending small molecule as quickly as possible if the patient is comatose or semicomatose.
5. Provide therapy specific to the disease; for example:
a. Nutritional modification, such as appropriate caloric supplements free of the offending precursor nutrients (eg, leucine, isoleucine, and valine in MSUD)
b. Cofactor administration, which will sometimes improve the function of a genetically defective enzyme (eg, vitamin B12 in some cases of methylmalonic aciduria, because methylmalonic acid is a cofactor for methylmalonyl-CoA mutase)
c. Metabolic manipulation, such as administering sodium benzoate in hyper-ammonemias to divert a toxic substrate to a benign excretable form
Later Onset, Acute, and Recurrent Attacks (Late Infancy and Beyond)
In about 50% of the patients with inborn errors of intermediary metabolism, onset of symptoms is delayed. The symptom-free period is often longer than 1 year and may extend into late childhood, adolescence, or even adulthood. Each attack can follow a rapid course ending either in spontaneous improvement or unexplained death despite supportive measures. Between attacks, the patient may appear normal. Onset of acute disease may occur without overt cause but may be precipitated by an intercurrent event related to excessive protein intake, prolonged fasting, prolonged exercise, or any condition that enhances protein catabolism.
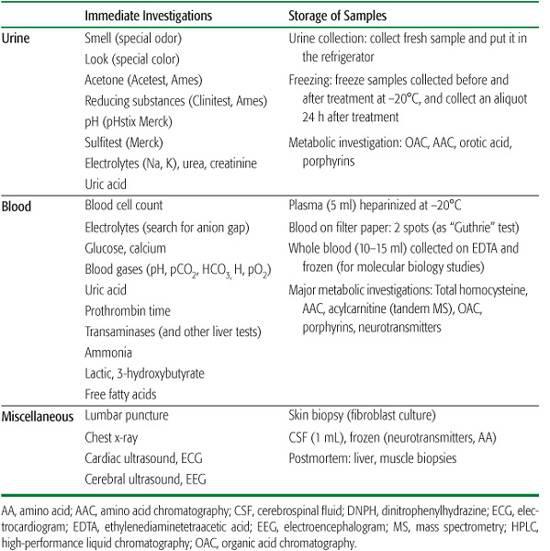
Table 134-3. Protocol for Emergency Investigations (Neonatal and Late-Onset Situations)
 Coma, Strokes, and Attacks of Vomiting with Lethargy
Coma, Strokes, and Attacks of Vomiting with Lethargy
Acute encephalopathy is a common problem in children and adults with IEM. All types of coma may be indicative of an IEM, including those presenting with focal neurological signs (Table 134-5). Neither the age at onset, the accompanying clinical signs (hepatic, gastrointestinal, neurological, psychiatric, etc), the mode of evolution (improvement, sequelae, death), nor the routine laboratory data allow an IEM to be ruled out a priori. Two categories can be distinguished:
1. Metabolic coma without focal neurological signs. The main varieties of metabolic coma may be observed in these late-onset, acute diseases, such as predominant metabolic acidosis, predominant hyperammonemia, predominant hypoglycemia, or combinations of these three abnormalities. A rather confusing finding in some organic acidurias and ketolytic defects is ketoacidosis with hyperglycemia and glycosuria that mimics diabetic coma. The diagnostic approach to these metabolic derangements is discussed below.
2. Neurological coma with focal signs, seizures, severe intracranial hypertension, strokes, or strokelike episodes. Although most recurrent metabolic comas are not accompanied by neurological signs other than encephalopathy, some patients with organic acidemias and urea cycle defects present with focal neurological signs or cerebral edema. These patients can be mistakenly diagnosed as having a cerebrovascular accident or cerebral tumor. In these disorders, stopping the protein intake, infusing large amounts of glucose, and giving “cleansing drugs” (carnitine, sodium benzoate, etc) can be lifesaving. Biotin-responsive basal ganglia disease is a treatable condition that presents in childhood with a subacute encephalopathic picture of undefined origin, including confusion, vomiting, and a vague history of febrile illness.11-13
All severe forms of homocystinuria (total homocysteine >100 μM/L) can cause an acute cerebrovascular accident from late childhood to adulthood. These forms include cystathionine-β-synthase deficiency (usually B6-responsive in late-onset presentations), severe methylenetetrahydrofolate reductase (MTHFR) defects (folate responsive), and cobalamin defects CblC and CblD (hydroxocobalamin responsive). Patients with methylmalonic acidemia (MMA) may, after first presenting with metabolic decompensation, have acute extrapyramidal and corticospinal tract involvement caused by destruction of the globus pallidus bilaterally. Glutaric acidemia (GA) type I frequently presents with an encephalopathic episode, mimicking encephalitis. Mitochondrial encephalopathy lactic acidosis strokelike episodes (MELAS syndrome) is another important diagnostic consideration in such late-onset and recurrent comas. Early episodic central nervous system problems, possibly associated with liver insufficiency or cardiac failure, have been the initial findings in some cases of CDG syndrome. Wilson disease can rarely present with an acute episode of encephalopathy with extrapyramidal signs.
In summary, all these disorders should be considered in the differential diagnosis of strokes or strokelike episodes. Vaguely defined or undocumented diagnoses such as encephalitis, basilar migraine, intoxication, poisoning, or cerebral thrombophlebitis should therefore be questioned, particularly when even moderate ketoacidosis, hyperlactatemia, or hyperammonemia is present. In fact, these apparent initial acute manifestations are frequently preceded by other premonitory symptoms, such as acute ataxia, persistent anorexia, chronic vomiting, failure to thrive, hypotonia, and progressive developmental delay—all symptoms that are often observed with urea cycle disorders (eg, ornithine transcarbamylase deficiency [OTC] and argininosuccinic aciduria), respiratory-chain defects, late maple syrup urine disease (MSUD) and organic acidurias. Late-onset forms of pyruvate dehydrogenase (PDH) deficiency can present in childhood with recurrent attacks of ataxia, sometimes described by the patient as recurrent episodes of pain or muscular weakness (due to dystonia or to peripheral neuropathy). Hartnup disease (the molecular mechanism of which has been recently identified) is a classical but rare cause of acute recurrent ataxias.
When coma is associated with hepatic dysfunction, Reye syndrome secondary to disorders of fatty acid oxidation or the urea cycle should be considered. Hepatic coma with liver failure and hyperlactatemia can be the presenting sign of respiratory-chain disorders. Hepatic coma with cirrhosis, chronic hepatic dysfunction, hemolytic jaundice, and various neurological signs (psychiatric, extrapyramidal) is a classic but underdiagnosed manifestation of Wilson disease.
 Acute Psychiatric Symptoms
Acute Psychiatric Symptoms
Late-onset forms of congenital hyperammonemia, mainly partial OTC deficiency, can present late in childhood or in adolescence with psychiatric symptoms. Because hyperammonemia and liver dysfunction can be mild even at the time of acute attacks, these intermittent late-onset forms of urea cycle disorders can easily be misdiagnosed as hysteria, schizophrenia, or alcohol or drug intoxication. Acute intermittent porphyria and hereditary coproporphyria present classically with recurrent attacks of vomiting, abdominal pain, neuropathy, and psychiatric symptoms. Patients with homocysteine remethylation defects may present with schizophrenia-like episodes that are responsive to folate. In view of these possible diagnoses, it is justified to systematically measure ammonia, porphyrins, and plasma homocysteine in every patient presenting with unexplained acute psychiatric symptoms.
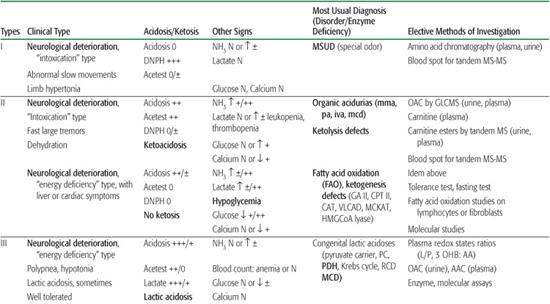
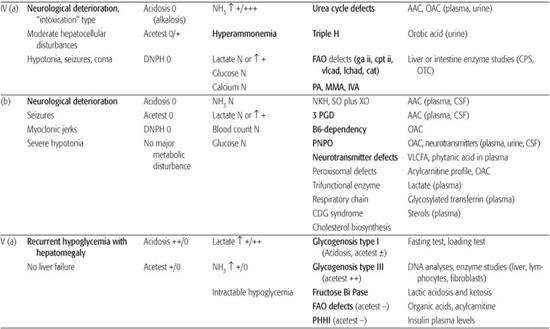
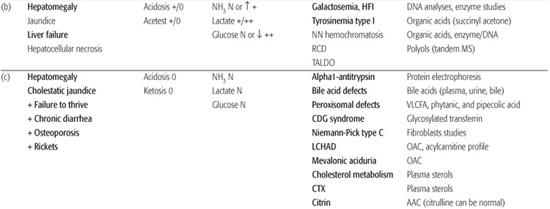
Table 134-4. Classification of Inborn Errors Revealed in the Neonatal Period and Early in Infancy
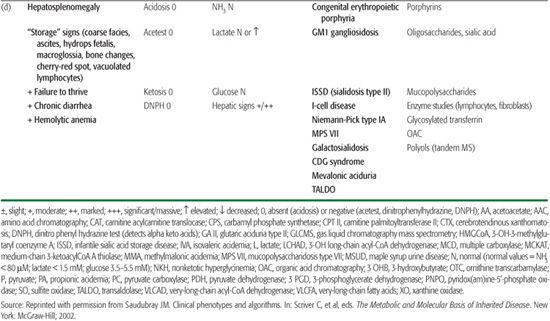
 Reye Syndrome, Sudden Unexpected Death in Infancy (SUDI), and Near-Miss
Reye Syndrome, Sudden Unexpected Death in Infancy (SUDI), and Near-Miss
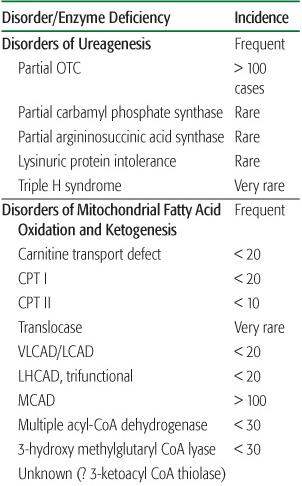
Within the last decade, an increasing number of IEM have been described that produce episodes fulfilling the criteria originally used to define Reye syndrome (Table 134-6). There is now considerable evidence that many of the disorders responsible for Reye syndrome were misdiag-nosed in the past because of inadequate investigations for IEM. Another important reason for this underestimation is that blood and urine specimens for metabolic investigations must be collected at an appropriate time in relation to the illness, because most conditions affecting the mitochondrial pathway and urea cycle and fatty acid oxidation (FAO) disorders may produce only intermittent abnormalities. In addition, a normal or nonspecific urinary organic acid and acylcarnitine pattern, even at the time of an acute attack, does not exclude an inherited FAO disorder. However, true SUDI due to an IEM is a rare event despite the large number of publications on the topic and despite the fact that at least 31 metabolic defects are possible causes. This assertion is not true in the first week of life, in which unexpected death (SIDS or near-miss) may be due to a fatty acid oxidation disorder. Investigation for this disorder is mandatory.
Table 134-5. Diagnostic Approach to Recurrent Attacks of Coma and Vomiting with Lethargy
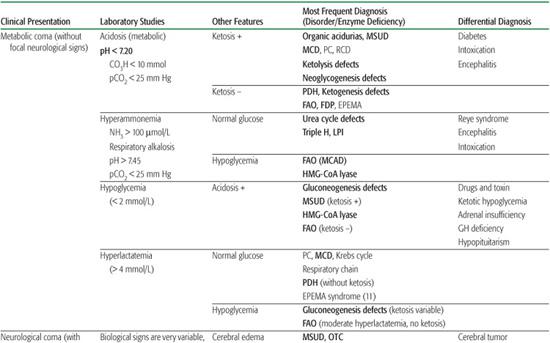
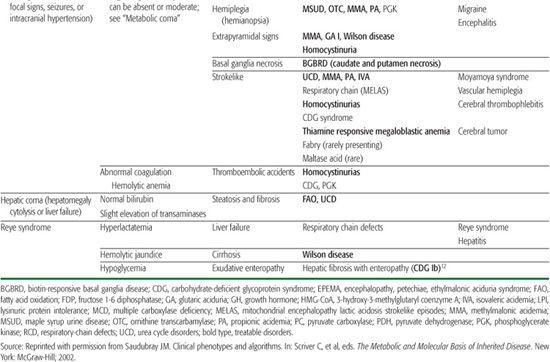
Table 134-6. Reye Syndrome, Sudden Unexpected Death in Infancy (SUDI) and Near-Miss SUDI
 Initial Approach to and Protocol for Investigation of Acute Late-Onset Encephalopathy
Initial Approach to and Protocol for Investigation of Acute Late-Onset Encephalopathy
As with the approach to acute neonatal distress, the initial approach to these disorders is based on the appropriate use of a few screening tests. As with neonates, the laboratory data listed in Table 134-7 must be collected simultaneously during the acute attack and before and after treatment.
Chronic and Progressive General Symptoms
Many late-onset acute presentations of IEM are actually preceded by premonitory symptoms that may have been ignored or misinterpreted. These symptoms fall schematically into three categories: gastrointestinal, muscular, or neurological.
 Gastrointestinal Involvement, Failure to Thrive, Anemia, and Recurrent Infections
Gastrointestinal Involvement, Failure to Thrive, Anemia, and Recurrent Infections
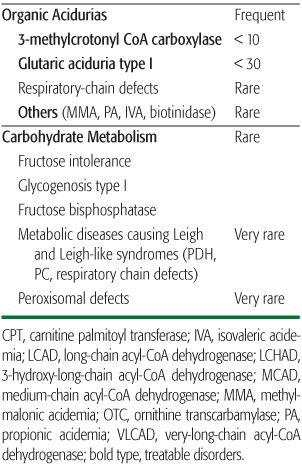
Gastrointestinal (GI) findings (anorexia, failure to thrive, chronic vomiting) and osteoporosis occur in a wide variety of IEM. Unfortunately, their cause often remains unrecognized, thus delaying the diagnosis. Persistent anorexia, feeding difficulties, chronic vomiting, failure to thrive, frequent infections, osteopenia, generalized hypotonia in association with chronic diarrhea, anemia, and bone marrow suppression are frequent presenting symptoms and signs in IEM. They are easily misdiagnosed as cow’s milk protein intolerance; celiac disease; chronic ear, nose, and throat infections; late-onset chronic pyloric stenosis, and so on. Congenital immunodeficiencies are also frequently considered, although only a few present early in infancy with this clinical picture. Faced with these presentations with no definitive diagnosis despite extensive gastroenterological, hematologic, and immunologic investigation, it is mandatory to seriously consider conditions such as organic aciduria-methylmalonic aciduria (MMA), proprionic acidemia (PA), isovaleric acidemia (IVA); urea cycle defects, ammonemia; lysinuric protein intolerance and respiratory-chain defects. Appropriate studies should be carried out.
 Muscle Involvement
Muscle Involvement
Many IEM present with severe hypotonia, muscular weakness, and poor muscle mass. These include most of the late-onset forms of urea cycle defects and many organic acidurias. Severe neonatal generalized hypotonia and progressive myopathy, with or without an associated nonobstructive idiopathic cardiomyopathy, can be the presenting features of mitochondrial respiratory-chain disorders and other congenital hyperlactatemias, FAO defects, peroxisomal disorders, muscular glycogenolysis defects, Pompe disease, and some other lysosomal disorders.
 Neurological Involvement
Neurological Involvement
Neurological symptoms are very frequent and encompass progressive psychomotor retardation, seizures, several neurological abnormalities in both the central and peripheral system, sensorineural defects, and psychiatric symptoms.
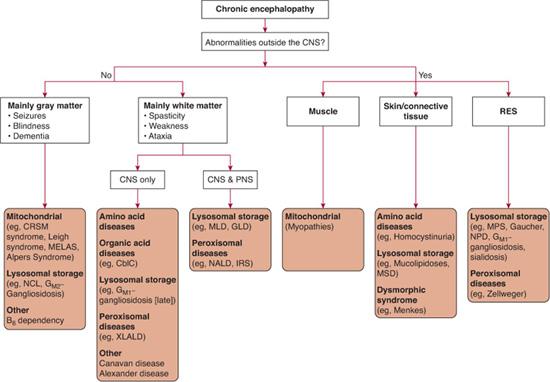
However, some aminoacidopathies and urine organic acidopathies that were identified in children with mental retardation in the late 1970s, when urine and plasma amino acid chromatography was first systematically measured, are now recognized to be of similar frequency in unaffected populations, such that their causative relationship with mental retardation is doubtful. These include histidinemia, hyperlysinemia, some types of hyperprolinemia, alpha-amino-adipic aciduria, saccharopinuria, and acetyl amino aciduria due to amino acylase I deficiency,14,15 adenylosuccinase deficiency, dihydropyrimidine dehydrogenase deficiency, 4-hydroxybutyric aciduria, D-2-hydroxyglutaric acidurias, and late-onset NKH. Several other inborn errors are now known to rarely, if ever, cause true developmental arrest. Rather, repeated episodes of subacute metabolic crises result in progressive developmental delay. An approach to identification of inborn errors of metabolism associated with chronic encephalopathy is shown in Figure 134-3.
Neurological signs of IEM can be classified according to age at presentation, the presence or absence of associated extraneurological signs, and the neurological presentation itself. IEM with neurological signs presenting in the neonate (birth to 1 month) and those presenting intermittently as acute attacks of coma, lethargy, ataxia, or acute psychiatric symptoms were discussed earlier.
Some IEM present with early and nonspecific progressive developmental delay, poor feeding, hypotonia, some degree of ataxia, and frequently autistic features. The list of conditions is rapidly getting longer. Many aminoacidopathies that were first described the late 1970s, when plasma and urine amino acid chromatography was systematically used in studying mentally retarded children, must now be questioned as causing neurological disease. This is the case for histidinemia, hyperlysinemia, some type of hyperprolinemia, alpha-amino-adipic aciduria, saccharopinuria, and acetyl amino aciduria due to amino acylase I deficiency.15 A similar picture is now emerging with organic acidurias. Among the new categories of inborn errors of intermediary metabolism that can present with uninformative clinical manifestations are, for example, adenylosuccinase deficiency, dihydropyrimi-dine dehydrogenase deficiency, 4-hydroxybutyric aciduria, D-2-hydroxyglutaric acidurias, late-onset NKH, and many other inborn errors. These disorders rarely, if ever, cause true developmental arrest; rather, they cause progressive subacute developmental delay.
Table 134-7. Diagnostic Approach to Inborn Errors of Metabolism with Chronic Diarrhea, Poor Feeding, Vomiting, Failure to Thrive
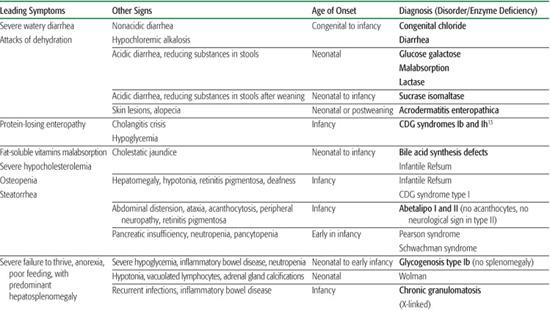
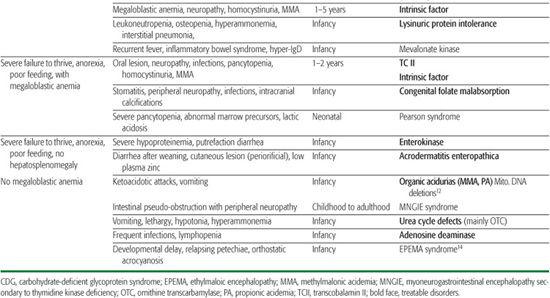
Early Infancy
Disorders Associated with Extraneurological Symptoms  Visceral signs appear in lysosomal disorders. Cardiomyopathy (associated with early neurological dysfunction, failure to thrive, and hypotonia), sometimes responsible for cardiac failure, is suggestive of respiratory-chain disorders, D-2-hydroxyglutaric aciduria (with atrioventricular block), or CDG syndrome. Abnormal hair and cutaneous signs appear in Menkes disease, Sjögren-Larsson syndrome, biotinidase deficiency, and respiratory-chain disorders. Peculiar fat pads on the buttocks, thick and sticky skin, and inverted nipples are highly suggestive of CDG syndrome. Generalized cyanosis unresponsive to oxygen, suggesting methemoglobinemia and associated with severe hypertonicity, indicates cytochrome-b5-reductase deficiency. Ortho-static acrocyanosis, relapsing petechiae, pyramidal signs, mental retardation, and recurrent attacks of lactic acidosis suggest ethylmalonic encephalopathy (EPEMA syndrome).16 The presence of megaloblastic anemia suggests an inborn error of folate and cobalamin (Cbl) metabolism. Ocular abnormalities, such as cherry-red spot, optic atrophy, nystagmus, abnormal eye movements, and retinitis pigmentosa, can be extremely helpful diagnostic signs
Visceral signs appear in lysosomal disorders. Cardiomyopathy (associated with early neurological dysfunction, failure to thrive, and hypotonia), sometimes responsible for cardiac failure, is suggestive of respiratory-chain disorders, D-2-hydroxyglutaric aciduria (with atrioventricular block), or CDG syndrome. Abnormal hair and cutaneous signs appear in Menkes disease, Sjögren-Larsson syndrome, biotinidase deficiency, and respiratory-chain disorders. Peculiar fat pads on the buttocks, thick and sticky skin, and inverted nipples are highly suggestive of CDG syndrome. Generalized cyanosis unresponsive to oxygen, suggesting methemoglobinemia and associated with severe hypertonicity, indicates cytochrome-b5-reductase deficiency. Ortho-static acrocyanosis, relapsing petechiae, pyramidal signs, mental retardation, and recurrent attacks of lactic acidosis suggest ethylmalonic encephalopathy (EPEMA syndrome).16 The presence of megaloblastic anemia suggests an inborn error of folate and cobalamin (Cbl) metabolism. Ocular abnormalities, such as cherry-red spot, optic atrophy, nystagmus, abnormal eye movements, and retinitis pigmentosa, can be extremely helpful diagnostic signs  .
.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


