Primary Immunodeficiencies
Talal A. Chatila
Primary immunodeficiency diseases result from genetic defects affecting the development and/or the function of immune system components.1-5 They frequently manifest soon after birth, although some become evident later in life. Recurrent infections are the hallmark of immunodeficiency syndromes, with the types of infections reflecting the nature of the defect. Autoimmune disorders and certain malignancies occur more frequently in individuals with immunodeficiency diseases.
Primary immunodeficiency diseases may affect either acquired immunity, as seen in cellular and humoral deficiency diseases, or innate immunity, such as seen in deficiencies of the various complement components and phagocytic cell deficiencies. This chapter reviews representative immunodeficiency diseases of altered development and function of T and B cells. These defects have served as experiments of nature to unravel the complexities of T-cell and B-cell development and function. T-cell immunodeficiencies resulting from defects affecting T-cell development and activation are shown in Figure 188-1, and B-cell immunodeficiencies due to abnormalities in B-cell development and differentiation are shown in Figure 188-2. Diseases of the innate immune system are reviewed in Chapter 187. Disorders of phagocytosis and other granulocyte disorders including Chediak-Higashi syndrome, cartilage-hair hypoplasia syndrome, myelokathexis, specific granule deficiency, leukocyte adhesion deficiency, chronic granulomatous disease and myeloperoxidase deficiency are discussed in Chapter 442.
Because of the role of the T lymphocyte in supporting B lymphocyte function, abnormalities affecting T cells alone or in combination with B lymphocytes frequently lead to states of combined immunodeficiency, the severity of which depends on the extent of the T-cell defect. In contrast to combined immunodeficiency diseases, where both T-cell and B-cell compartments are affected, T-cell function in isolated B-cell immunodeficiency syndromes, such as X-linked agammaglobulinemia, remains intact.
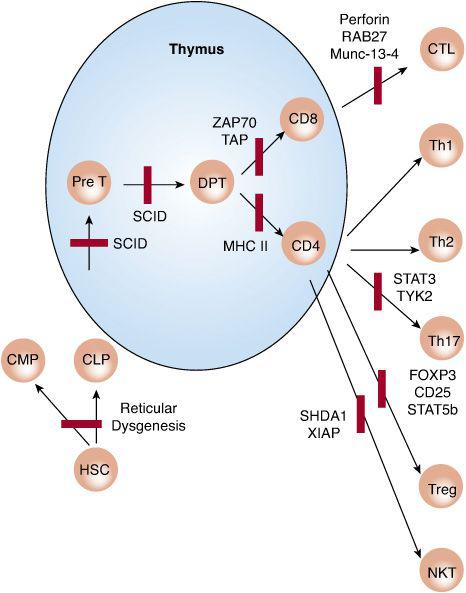
FIGURE 188-1. Schematic representation of T-cell immunodeficiencies resulting from defects affecting T-cell development and activation. Bars indicate a block in cell development or function. CLP and CMP, common lymphoid and myeloid progenitors, respectively; DPT, double-positive thymocytes; HSCs, hematopoietic stem cells; NKT, natural killer cell; PreT, pre-T cells; SCID, severe combined immunodeficiency.
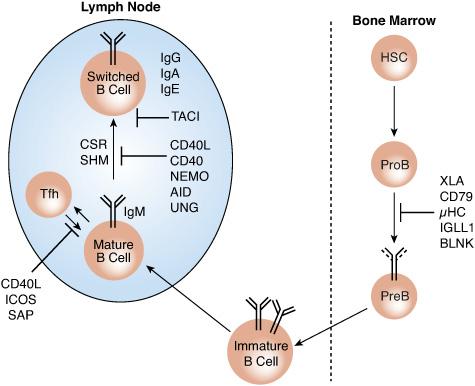
FIGURE 188-2. B-cell immunodeficiencies due to abnormalities in B-cell development and differentiation. AID, activation-induced cytidine deaminase; BLNK, B cell LINKer protein; CD, cluster of differentiation; CSR, class switch recombination; HSCs, hematopoietic stem cells; IGLL1, immunoglobulin lambda-like polypeptide 1; μHC, Ig membrane μ heavy chain; NEMO, NF-κB essential modulator; PreB, pre-B cells; ProB, pro-B cells; SHM, somatic hypermutation; TACI, transmembrane activator and CAML interactor; Tfh, T follicular helper B cells; UNG, uracil-DNA glycosylase; XLA, X-linked agammaglobulinemia.
SEVERE COMBINED IMMUNODEFICIENCY
Severe combined immunodeficiency (SCID) is characterized by the breakdown of both cellular and humoral adaptive immunity. The reported incidence is approximately 1 per 100,000.6,7 It is caused by a heterogeneous group of at least 15 known genetic abnormalities that result in severe T-cell depletion (or dysfunction) with either primary or secondary B-cell involvement. Recently, screening for SCID has been initiated in a few geographic regions. The thymus is usually very small and is depleted of thymocytes and Hassall corpuscles. Peripheral lymphoid tissues are commonly atrophied and depleted of lymphocytes. Severe combined immunodeficiency diseases are usually classified according to the presence or absence of lymphocyte subsets (T, B, and NK cells). For example, a patient with T−B−NK+ severe combined immunodeficiency (T−B−NK+ SCID) indicates that both the T- and B-cell populations are profoundly decreased or absent and NK cells are spared. Such a classification provides clues to the underlying genetic defects.
Severe combined immunodeficiency poses the risk of imminent death caused by infection with a wide variety of microorganisms, including bacteria, viruses, fungi, and protozoa. Persistent infections of the lung, chronic diarrhea, and wasting dominate the clinical picture. Candidal infections of the oropharynx, esophagus, and skin are common and are among the early manifestations of the disease. Infections with rotavirus may be persistent and may spread to extraintestinal sites, and respiratory syncytial virus infections may result in giant cell pneumonia. Infections with varicella, herpes, measles, and adenoviruses are frequently fatal. Patients with severe combined immunodeficiency are also susceptible to opportunistic infections, including Pneumocystis jirovecii and cytomegalovirus, which cause chronic, progressive pneumonitis. For patients with severe combined immunodeficiency, fatal infections may follow vaccination with live vaccine vectors such as measles or with the mycobacterium Bacillus Calmette-Guerin. Transfusion with blood that has not been irradiated may lead to severe graft-versus-host disease.
Most children with severe combined immunodeficiency require urgent medical attention within months after birth. However, the age at presentation and the clinical manifestations of severe combined immunodeficiency may vary among different patients, including those with the same disease etiology and even between affected members of the same family. Depending on the underlying abnormality, some patients with severe combined immunodeficiency may begin life with a seemingly normal immune function only to subsequently suffer progressive immunodeficiency.
 T−B− SEVERE COMBINED IMMUNODEFICIENCY
T−B− SEVERE COMBINED IMMUNODEFICIENCY
Reticular Dysgenesis
T−B−SCID, a rare form of severe combined immunodeficiency, is characterized by failure of lymphoid and myeloid lineage development in the face of normal erythroid and megakaryocytic cell development. It is caused by mutations in the gene encoding mitochondrial adenylate kinase 2 (see Fig. 188-1).8 Clinically, affected neonates exhibit alymphocytosis and agranulocytosis and sensorineural deafness. The disease is fatal in infancy unless treated with bone marrow transplantation, which is curative.
Defects Affecting T and B Lymphocyte Antigen Receptor Rearrangements
T−B−NK+ SCID caused by failure of antigen receptor rearrangement are a group of autosomal recessive disorders resulting from mutations and deletions in genes involved in the diversification of T- and B-cell receptors. They are characterized by the profound decrease or absence of T and B cells at all stages of development. In contrast, NK cells are typically spared in these disorders because they do not undergo antigen receptor rearrangement and are governed by distinct developmental pathways.
The process of antigen receptor diversification involves the generation of double-stranded DNA breaks during V(D)J recombination by the action of the protein products encoded by the lymphoid-specific recombination activating genes (RAG) 1 and 2. The DNA breaks are subsequently repaired by the action of a ubiquitous DNA repair mechanism known as nonhomologous end joining. The RAG genes cooperate in initiating V(D)J recombination in T and B lymphocytes, and deficiency of either gene product abrogates this reaction and gives rise to T−B−SCID.9 Mutations affecting components of the nonhomologous end joining machinery, including the DNA cross-link repair 1C (DCLRE1C, ARTEMIS) and nonhomologous end-joining factor 1 (NHEJ1, CERNUNNOS), also result in a T−B−NK+ SCID phenotype. Mutations affecting DNA ligase IV gene LIG4, which is a component of the nonhomologous end joining machinery, give rise to a variable phenotype ranging from mild immunodeficiency to T−B− SCID.
Because the RAG genes are selectively expressed in lymphoid tissues, their deficiency is not associated with extralymphoid manifestations. However, the manifestations of gene defects involving nonhomologous end joining extend beyond the immune system to include broad radiation sensitivity (due to inability to repair radiation-induced DNA breakage in different tissues). Some nonhomologous end joining defects, such as those affecting Cernunnos and DNA ligase IV, also result in microcephaly.
Omenn syndrome is a variant form of RAG1/2 and ARTEMIS deficiency resulting from hypomorphic (partially inactivating) mutations affecting these genes.10 Clinically, patients with this disorder present with intense erythroderma, protracted diarrhea, hepatosplenomegaly, and failure to thrive. Investigation typically reveals hypogammaglobulinemia, elevated IgE, and intense eosinophilia with T-cell infiltration of several organs, including the skin, gut, liver, and spleen. There is severe lymphocyte depletion in the thymus and other lymphoid organs. B cell numbers are usually profoundly depressed. The T cells may be present in the peripheral blood in decreased, normal, or even elevated numbers; however, their heterogeneity is severely restricted because they represent the expansion of a small number of T-cell clones that have survived the aberrant thymic development. Those surviving T-cell clones are skewed toward Th2 phenotype, consistent with the picture of high IgE and eosinophilia. Siblings of patients with Omenn syndrome may present with a complete T−B−NK+ SCID picture, consistent with a variable penetrance of the same genetic defect in different family members.11
 SEVERE COMBINED IMMUNODEFICIENCY DUE TO DISORDERS OF THE PURINE SALVAGE PATHWAY
SEVERE COMBINED IMMUNODEFICIENCY DUE TO DISORDERS OF THE PURINE SALVAGE PATHWAY
Two specific disorders of the purine salvage pathway, involving the adenosine deaminase (ADA) gene and purine nucleoside phosphorylase (PNP) gene deficiency, account for 15% to 20% of all severe combined immunodeficiency cases. Adenosine deaminase deficiency, by far the more common of the two defects, compromises the viability of both T and B cells and is particularly toxic to developing thymocytes. In contrast, purine nucleoside phosphorylase deficiency more selectively impacts T cells and their progenitors, although the B cells may also be affected. Both conditions are inherited in an autosomal recessive form.
Adenosine deaminase catalyzes the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine, respectively. Its deficiency results in the accumulation in lymphocytes of deoxyadenosine and a metabolite, deoxyadenosine triphosphate, that is particularly toxic to lymphocytes, especially thymocytes. Within months after birth, patients with adenosine deaminase deficiency usually present with recurrent infections associated with profound lymphopenia and hypogammaglobulinemia. However, they may also present with progressive immunodeficiency, with the results of initial studies showing normal or near normal immune parameters. Skeletal abnormalities, including cupping and flaring of the costochondral junctions and dysplasia of the pelvis are frequent but not pathognomonic. The thymus is depleted of thymocytes. Diagnosis is established by demonstrating low adenine deaminase activity in blood cells in conjunction with elevated levels of deoxyadenosine and deoxyadenosine triphosphate in blood and urine. Prenatal diagnosis is possible using fetal blood samples. In all cases, the outcome is fatal unless treatment is provided. Bone marrow transplantation cures this disease.12 Enzyme replacement therapy with ethylene glycol–modified adenosine deaminase provides an alternative treatment modality that is safe and relatively effective for those patients lacking a suitable bone marrow donor.13
Unlike adenosine deaminase deficiency, in purine nucleoside phosphorylase (PNP) deficiency T cell numbers and function are profoundly depressed but there may be relatively normal B cell numbers and immunoglobulin levels.14 PNP catalyzes the conversion of inosine to hypoxanthine and guanosine to guanine. PNP deficiency results in the accumulation of the toxic metabolites deoxyguanosine triphosphate and deoxyinosine triphosphate. In addition to recurrent infections, about one third of the patients suffer from neurologic abnormalities and autoimmunity, particularly autoimmune hemolytic anemia and thrombocytopenia. Bone marrow transplantation is currently the only therapeutic modality available for these children. Diagnosis is established by demonstrating low PNP activity in blood cells in conjunction with elevated levels of deoxyguanosine and deoxyguanosine triphosphate in blood and urine.
 T−B+ SCID
T−B+ SCID
X-Linked Severe Combined Immunodeficiency, JAK3 and IL-7 Receptor Deficiency
Severe combined immunodeficiency (SCID) with selective failure of T and NK cell development in the face of normal or increased peripheral B cell numbers (T−B+NK− SCID) accounts for up to 25% of all SCID patients. Most of those cases are males with an X-linked form of SCID. However, autosomal recessive forms related in pathogenesis to the X-linked SCID variety have been described. As is detailed below, the X-linked and autosomal recessive forms result from distinct molecular defects affecting a common cytokine signaling pathway essential for T-cell development in the thymus.
X-linked severe combined immunodeficiency is characterized by the failure of T and NK cell development and the absence of host T cells from circulation and from lymphoid organs. The thymus is hypoplastic and is devoid of thymocytes and Hassall corpuscles. Although trans-placental engraftment of maternal lymphocytes frequently results in the presence of significant numbers of circulating T cells, these maternal T cells display very poor function. The numbers of circulating B cells are usually increased and their phenotype is normal, indicating that the defect does not significantly interfere with B cell development. However, the B cells of children with X-linked severe combined immunodeficiency exhibit abnormal function. They produce reduced amounts of immunoglobulins in vitro and appear to suffer from a block affecting their terminal differentiation.
Family history consistent with X-linked disease is present in one third of the patients. Most of the remaining cases result from an unrecognized maternal carrier state or spontaneous mutations. T cells of female carriers display skewed (nonrandom) inactivation of their X chromosome. Their T cells appear to retain the X chromosome carrying the wild-type but not the mutant allele, indicating an essential role for IL2RG in T-cell survival.
Immunodeficiency in X-linked severe combined immunodeficiency results from mutations in IL2RG. It encodes the common cytokine receptor γ-chain subunit, also termed γc and CD132, which plays an essential role in signaling via several cytokine receptors, including those for IL-2, IL-4, IL-7, IL-9, and IL-15. Although selective disruption of other γc-containing cytokine receptors is associated with distinct forms of immunodeficiency, it appears that the total failure of T-cell development in X-linked severe combined immunodeficiency is related to absent IL-7 receptor function.15 This was revealed by the discovery of severe combined immunodeficiency cases with selective T-cell deficiency resulting from inactivating mutations in the IL-7 receptor α-chain gene (IL7R). NK cells are spared in this form of severe combined immunodeficiency, reflecting the requirement for the IL-15, but not the IL-7, receptor in NK cell development.
A second type of autosomal recessive T−B+NK–-deficient severe combined immunodeficiency results from mutations in the gene encoding the Janus-type protein tyrosine kinase JAK3, which associates with the γc chain and mediates its signaling function.16 The clinical phenotype of JAK3 deficiency is identical to that of X-linked severe combined immunodeficiency, with depletion of both T and NK cells. Taken together, the three defects (γc, JAK3, and IL-7 receptor) elegantly demonstrate how overlapping severe combined immunodeficiency phenotypes result from the disruption of sequential steps along a signal transduction pathway.
Diagnosis of X-linked severe combined immunodeficiency and related disorders is accomplished by examining T lymphocytes of carrier females for nonrandom X chromosome inactivation and/or by direct sequencing of affected genes (IL2RG and, when indicated, JAK3 and IL7R). Bone marrow transplantation restores normal T-cell development and function. However, it frequently fails to correct the impaired B-cell function in X-linked severe combined immunodeficiency and JAK3 deficiency because the donor B cells often fail to engraft. In such cases, intravenous immunoglobulin therapy is maintained indefinitely.
 CD3δ/ε/ζ AND CD45 DEFICIENCIES
CD3δ/ε/ζ AND CD45 DEFICIENCIES
Expression of the T-cell antigen receptor on the surface of T cells and the generation of downstream activation signals is critically dependent on its association with a number of subunits collectively known as the CD3 complex. The minimal T-cell receptor (TCR)/CD3 complex has eight subunits with a stoichiometry of one TCR-α/β heterodimer, one CD3-ε/g heterodimer, one CD3-ε/δ heterodimer, and a CD3-ζ homodimer. Mutations in the TCR-associated CD3 subunits δ, ε, and ζ result in a block in T-cell development in the thymus and a phenotype of T−B+NK+ SCID.17 CD3γ chain deficiency results in a milder phenotype with normal or decreased circulating T cells in the periphery, but with decreased surface expression of TCR/CD3.
Signaling via TCR/CD3 complex is dependent on the protein tyrosine phosphatase CD45, which enables repetitive signaling by the TCR/CD3 complex by resetting the signaling back to baseline between bouts of activation.18 CD45 deficiency in two unrelated cases resulted in a phenotype of T−B+NKM+ SCID, mirroring previous observations made in mouse models of this disorder.
PRIMARY T AND COMBINED T AND B IMMUNODEFICIENCIES
 MHC CLASS I AND CLASS II DEFICIENCIES
MHC CLASS I AND CLASS II DEFICIENCIES
Patients failing to express MHC class I or class II molecules present with specific abnormalities affecting the development and functioning of CD8+ and CD4+ cells, respectively (see Fig. 188-1). In MHC class II deficiency, the development of CD4+ T cells in the thymus is markedly impaired because of their inefficient positive selection. The available CD4+ T cells cannot mount delayed-type hypersensitivity responses nor can they provide help to B cells to generate antigen-specific humoral responses, resulting in a combined immunodeficiency. Responses to mitogenic lectins and to allogeneic cells are usually preserved. Patients with MHC class II deficiency suffer from recurrent infections similar to those seen in severe combined immunodeficiency. In addition, they are prone to autoimmunity, a direct consequence of ineffective negative selection of CD4+ T cells in the thymus.
MHC class II deficiency is an autosomal recessive disease that results from the failure to express MHC class II molecules, including HLA-DP, HLA-DQ, and HLA-DR. It is most prevalent in patients of Mediterranean ancestry. The underlying abnormalities resides not in the MHC genes, but in trans-acting regulatory factors that control the expression of these genes, including CTII-A, RFX-5, RFX-AP, and RFX-ANK.19
A reciprocal scenario of impaired CD8+ T-cell development and function attends MHC class I deficiency. These patients suffer from recurrent viral infections, and some are particularly prone to chronic lung disease. MHC class I deficiency results from autosomal recessive mutations in components of the pathway involved in loading antigenic peptides for presentation. MHC class I expression depends on the formation in the endoplasmic reticulum of a peptide-loading complex composed of MHC class I heavy chain; β2-microglobulin; the transporter associated with antigen processing (TAP), and the TAP binding protein tapasin (TAPBP), which links TAP to the MHC class I heavy chain. TAP is a heterodimeric transporter complex that translocates peptides from the cytosol to the endoplasmic reticulum. Mutations affecting either of the TAP subunits, TAP1 or TAP2, as well as TAPBP, impair peptide loading, resulting in unstable MHC class I/β2 microglobulin complexes that are inefficiently transported to the surface.
 ZAP70 DEFICIENCY
ZAP70 DEFICIENCY
The protein tyrosine kinase ZAP70 is selectively expressed in T lymphocytes and is activated on engagement of the T-cell receptor by antigen/MHC complexes (see Fig. 188-1). ZAP70 is indispensable to the program of T-cell activation, and its deficiency renders T cells unresponsive to antigenic stimulation. ZAP70 deficiency is an autosomal recessive disease that results from loss of function mutations in the kinase gene. It presents with recurrent infections and impairment of both cellular and humoral immunity.21,22 Patients have virtually no CD8+ T cells in circulation or in peripheral lymphoid organs, and the CD4+ T cells are poorly responsive to antigens or to mitogens. The thymus contains CD4+CD8+ double-positive immature thymocytes but no CD8+ single-positive mature thymocytes. Although the patients have normal numbers of CD4+ T cells, these cells do not respond to T-cell mitogens that act by engaging the T-cell receptor. Bone marrow transplantation successfully cures this condition.
 CALCIUM CHANNEL DEFICIENCY
CALCIUM CHANNEL DEFICIENCY
A rare cause of combined immunodeficiency is a mutation in Orai-1, a pore subunit of the calcium release–activated calcium channel, which mediates the influx of extracellular calcium that follows antigen receptor engagement. The circulating lymphocytes are normal in number and in phenotype, but the proliferative responses to mitogens are severely defective. In addition to the immunodeficiency, the patients also suffer from anhydrotic ectodermal dysplasia and a nonprogressive myopathy.
 DISORDERS AFFECTING THE THYMIC MICROENVIRONMENT
DISORDERS AFFECTING THE THYMIC MICROENVIRONMENT
The chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome is characterized by defective T-cell development due to the absence or disruption of the thymic microenvironment. It results from the impaired embryogenesis of a group of cephalic neural crest cells, leading to aberrant development of several tissues, including thymus, heart, and parathyroid gland.22 Most patients with this syndrome exhibit a 3-Mb hemizygous interstitial deletion in chromosome 22q11, and a small percentage present with chromosome 22 aneuploidy. Isolated haploinsufficiency of T-box factor 1, a transcription factor found in the deleted region, gives rise to an overlapping clinical phenotype. In the past, the 22q11 deletion syndrome was referred to as the DiGeorge syndrome; Shprintzen syndrome; velocardiofacial syndrome; or cleft palate, absent thymus, congenital heart disease syndrome. Diagnosis is possible by performing tests using fluorescent in situ hybridization and appropriate DNA probes. Currently, this condition is most properly referred to as the 22q11 deletion syndrome. Clinical manifestations of these disorders vary as discussed in Chapter 174. Although most cases are not inherited, vertical transmission of 22q11 deletions is well recognized.
Several conditions mimic the DiGeorge syndrome in the absence of 22q11 deletions, including the fetal alcohol syndrome and retinoic acid embryopathy. The winged helix nude (WHN) transcription factor is also required for normal development of thymic epithelium and hair follicles. Patients with WHN deficiency display the so called “nude” phenotype characterized by a severe decrease in circulating T cells and congenital alopecia and nail dystrophy.
In the patient with 22q11.2 deletion syndrome the thymus may be absent or, more commonly, hypoplastic. The spectrum of immune dysfunction ranges from the severe (so-called complete DiGeorge syndrome) to the subtle (partial DiGeorge syndrome). In the complete form of the DiGeorge syndrome that occurs in <0.5% of patients with 22q11.2 deletion syndromes, there exists a picture of T−B+NK+ SCID, with severe or total depletion of peripheral T cells.23 The B-cell numbers are normal or elevated, but hypogammaglobulinemia and absent antigen-specific antibody responses are prevalent due to absent T-cell help. Far more common is the picture of partial DiGeorge syndrome, in which T-cell depletion is moderate to minimal and B-cell number and function are normal.23 NK cell activity is generally unaffected in both forms of the disease. Defective thymic selection processes may predispose patients to autoimmune diseases, including juvenile rheumatoid arthritis. This may reflect disease-related defects in thymic selection, leading to the escape of autoreactive T cells into the periphery.
In most cases of partial DiGeorge syndrome, the immunologic deficit ameliorates with time because of compensatory residual thymic tissue or extrathymic maturation of T cells. The complete form of the syndrome has been successfully managed with thymic transplant. Intravenous immunoglobulin replacement therapy is beneficial in selected cases.
COMBINED IMMUNODEFICIENCIES ASSOCIATED WITH OTHER DEFECTS
In addition to the diseases of T-cell development and activation, there are a host of other diseases that are associated with T-cell dysfunction. Two diseases of particular interest are ataxia telangiectasia and the Wiskott-Aldrich syndrome.
 ATAXIA TELENGIECTASIA AND OTHER DISORDERS OF DNA REPAIR
ATAXIA TELENGIECTASIA AND OTHER DISORDERS OF DNA REPAIR
Ataxia telangiectasia (AT) is an autosomal recessive disease characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, immunodeficiency, high incidence of cancer, and increased sensitivity to ionizing radiation.24 A hallmark of ataxia-telangiectasia is heightened sensitivity to ionizing radiation and radio-mimetic agents, consistent with defective repair of damaged DNA as a major pathogenic feature of this disease. The responsible gene, ATM (AT–mutated), encodes a member of the phosphatidyl-inositol-3 kinase family that monitors DNA repair and coordinates DNA synthesis with cell cycle progression. ATM mutations impair protein expression and function, leading to the accumulation of DNA double-stranded breaks, the activation of programmed cell death (apoptosis), and the progressive depletion of affected cell types.
Ataxia manifests first as a staggering gait; its onset heralds other neurologic abnormalities, including ocular apraxia and choreoathetosis. Telangiectasias begin as dilatation of small blood vessels in the bulbar conjunctivae and become visible in the skin by age 5 years. They are most notable around the ears, the neck, and the antecubital and popliteal fossae. Endocrine abnormalities such as gonadal dysgenesis and insulin-resistant diabetes mellitus are frequent. Recurrent sinopulmonary infections reflect the underlying immunodeficiency and may lead to bronchiectasis. The serum levels of oncofeto-proteins, including serum a1-fetoprotein and carcinoembryonic antigen, are elevated. For children age 1 year or older, a serum a1-fetoprotein determination is the most sensitive test widely available, which, when coupled with the clinical picture, confirms the diagnosis. Recently, new rapid diagnostic tests based upon flow cytometry monitoring of DNA damage have been reported to identify both homozygotes and heterozygotes more rapidly and conveniently.25,26 This test is not yet widely available.
Immunodeficiency in ataxia telangiectasia reflects both cellular and humoral immune defects. The thymus is abnormally small and sparsely populated, and peripheral lymphoid tissues exhibit depletion of resident T cells. The number of circulating CD4+ T cells may be reduced, and T-cell functional responses are defective. B cells are found in normal numbers; however, there is an immunoglobulin isotype switch defect with profound IgA and IgE deficiency. IgG deficiency, found in up to one half of all patients, reflects a selective decrease in the IgG2 and IgG4 subclasses, whereas IgM levels are usually normal or elevated. Those patients with no ATM kinase activity appear to have more severe immunologic deficiencies than those with low levels of ATM activity.27 Immunoglobulin replacement therapy is indicated in patients with frank hypogammaglobulinemia and recurrent infections.26
Patients with ataxia telangiectasia are at a high risk for developing malignancies, particularly those of the lymphoid system, and tumors of epithelial origin increase progressively with age. Otherwise healthy disease carriers also have increased frequency of tumors. Breaks in chromosomes 7 and 14 at sites of rearranging T-cell receptors and immunoglobulin heavy-chain genes, respectively, contribute to tumorigenicity. Because of their heightened sensitivity to ionizing radiation and their increased risk for developing malignancies, exposure of patients with ataxia telangiectasia to x-rays and radiomimetic agents should be strictly curtailed.
Ataxia telangiectasia–like disorders result from mutations in genes, including NBN and MRE11, encoding components of a protein complex downstream from ATM that is involved in repair of DNA double-stranded breaks.28 These diseases are associated with impaired double-stranded DNA breakage and increased sensitivity to ionizing radiation. Mutations in NBN give rise to the Nijmegen breakage syndrome, a rare autosomal recessive disorder associated with microcephaly with normal or near normal intelligence, combined cellular and humoral immunodeficiency, and high incidence of malignancy, especially lymphoid malignancies.29 Mutations in MRE11 result in a milder phenotype.
 WISKOTT-ALDRICH SYNDROME
WISKOTT-ALDRICH SYNDROME
Wiskott-Aldrich syndrome is an X-linked immunodeficiency disease characterized by a triad of thrombocytopenia with small platelets, eczema, and increased susceptibility to pyogenic and opportunistic infections.30,31 A related disorder, X-linked thrombocytopenia, presents with isolated chronic thrombocytopenia. Between the two phenotypes are attenuated forms of Wiskott-Aldrich syndrome in which eczema and immunodeficiency are variably expressed. All three phenotypes result from mutations affecting the WASP (Wiskott-Aldrich syndrome protein) gene. WASP is a cytoplasmic proline-rich protein that integrates diverse signal transduction pathways and their interface with the cellular cytoskeleton. Mutations that severely compromise WASP expression lead to classical Wiskott-Aldrich syndrome, whereas milder mutations underlie X-linked thrombocytopenia and attenuated forms of the syndrome.32
Affected boys usually present with bleeding early in infancy. Eczema is frequently accompanied by eosinophilia, elevated IgE, and positive prick test reactions to common allergens, suggesting an underlying allergic etiology. The eczematous skin may become infected with Staphylococcus aureus. Immunodeficiency may manifest as persistent sinopulmonary infections and/or unusually severe childhood viral infections, including varicella. Wiskott-Aldrich syndrome patients are also at high risk of developing lymphomas and leukemias. Lymphomas commonly develop at extranodal sites, particularly in the brain and in the gastrointestinal tract. Overall, many Wiskott-Aldrich syndrome patients die due to bleeding, infection, or malignancy within their first decade of life.
The platelet numbers are invariably low and of small size in Wiskott-Aldrich syndrome which is a diagnostically useful feature. They display impaired aggregation in response to ADP, epinephrine, risotectin, and collagen. Thrombocytopenia is a consequence of both ineffectual thrombocytosis and, more importantly, enhanced sequestration due to abnormal platelet cytoarchitecture. Splenectomy ameliorates the thrombocytopenia and normalizes the reduced platelet volume in Wiskott-Aldrich syndrome patients. Autoimmune thrombocytopenia may complicate the course of some of these patients and precipitate postsplenectomy thrombocytopenia.
Immunodeficiency in Wiskott-Aldrich syndrome results from defects in both cellular and humoral immunity. T-cell responses progressively diminish with age, and cutaneous anergy is common. A characteristic morphological abnormality, a near absence of microvilli, is apparent when viewed with electron microscopy. Humoral abnormalities reflect, in part, the abnormalities in T-cell function. Typically, the serum IgA and IgE levels are elevated, the IgM decreased, and the IgG normal. Immune response to polysaccharide antigens is absent or very diminished; isohemagglutinins are absent; and the response to unconjugated polysaccharide vaccines is very poor. Responses to protein antigens are also defective, reflecting abnormal T-helper-cell function.
Obligate female heterozygotes exhibit non-random X chromosome inactivation in all blood cell lines, but not in other tissues such as fibroblasts, indicating that hematopoietic cells carrying the mutant WAS are impaired in their development and survival.
The treatment of choice in Wiskott-Aldrich syndrome is a bone marrow transplant from an HLA-matched donor. When a donor is not available, splenectomy often has a satisfactory outcome in improving the platelet count and normalizing platelet size. However, splenectomy compounds the immunologic deficit and increases the risk of overwhelming infections with encapsulated organisms. Immunoglobulin replacement therapy is beneficial in preventing pyogenic infections and is indicated especially after a splenectomy.31
 THE HYPER IGE SYNDROME
THE HYPER IGE SYNDROME
The hyper-IgE syndrome (HIES), also known as Job syndrome, is a rare immunodeficiency characterized by eczema, skeletal and connective tissue abnormalities, Staphylococcus aureus skin abscesses, pneumonia with abscess and pneumatocele formation, and candida infections.33 Inflammatory responses are characteristically aberrant in that infections cause significant tissue destruction but do not generate the expected warmth, redness, and fever. Immunological abnormalities include markedly elevated IgE levels, abnormal antibody responses, defective neutrophil chemotaxis, and altered cytokine expression patterns.
Hyper-IgE syndrome presents in an autosomal dominant (AD-HIES) as well as an autosomal recessive (AR-HIES) form. The autosomal dominant form is characterized by the presence of skeletal and mesenchymal tissue manifestations, including retention of decidual teeth, recurrent fractures, pneumatocele formation, and coronary vascular aneurysms. Up to half of the affected subjects with autosomal dominant hyper-IgE syndrome suffer from heterozygous mutations in the transcription factor signal transducer and activator of transcription 3 (STAT3). The mutations, which cluster in the DNA binding and SH2 domains, spare STAT3 protein expression but impair its activation, dimerization, and DNA binding. Many of the immunological abnormalities in autosomal dominant hyper-IgE syndrome, including the impaired inflammatory response, neutrophil chemotaxis, and perhaps the infection susceptibility profile, can be attributed to the failure of TH17-type T-helper-cell differentiation, which normally proceeds in a STAT3-dependent fashion.34 Autosomal recessive hyper-IgE syndrome lacks the skeletal and mesenchymal tissue manifestations, but instead is notable for recurrent viral infections with herpes simplex, varicella, and molluscum contagiosum virus. There is also a predilection for autoimmunity. The genetic lesions underlying most cases of autosomal recessive hyper-IgE syndrome remain unknown. However, one subject with an autosomal recessive hyper-IgE syndrome-like phenotype was found to have mutations in the gene encoding the protein tyrosine kinase TYK2, which locates upstream from STAT3 and other pathways involved in the innate and acquired immune responses.
PRIMARY ANTIBODY DEFICIENCY DISEASES
Patients with antibody deficiency syndromes usually present with a history of recurrent pyogenic infections of the respiratory tract or other organs and chronic gastrointestinal disease, including giardiasis. The bacterial infections are mainly due to encapsulated, pyogenic organisms such as Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, and Neisseria meningitidis. Otherwise intact cell-mediated immunity allows viral infections to be cleared, although they are prone to recur given absent protective antibody responses. Tonsils are absent in patients with agammaglobulinemia. Radiological studies may be helpful in evaluating the size of the adenoid tissue and in assessing the sinuses and lung fields. Evidence for recurrent infections, such as scars from previous abscesses or chronic otitis media, are often noted.
Measurement of serum immunoglobulins and specific antibody responses are critical screening tests in the diagnosis of antibody deficiencies. The determination of serum antibody titers after tetanus or diphtheria immunizations or immunization with Haemophilus influenzae and pneumococcal vaccines provides valuable information about the individual’s ability to mount specific antibody responses. When a patient is found to have low immunoglobulin levels, additional in vitro tests are used to characterize the nature of the B-cell defect, including enumeration and surface phenotyping of circulating B lymphocytes and in vitro immunoglobulin production assays.
 AGAMMAGLOBULINEMIA DUE TO DEFECTS IN EARLY B-CELL DEVELOPMENT
AGAMMAGLOBULINEMIA DUE TO DEFECTS IN EARLY B-CELL DEVELOPMENT
Rare genetic abnormalities affecting early B-cell development result in deficiency of mature B cells, leading to agammaglobulinemia. Prevalence is about 1 per 100,000. Most (85%) of the affected subjects are males who suffer from X-linked agammaglobulinemia (XLA), and the remainder are both males and females suffering from a heterogeneous group of autosomal recessive disorders.35,36
XLA is characterized by profoundly low serum concentrations of all immunoglobulin classes and the virtual absence of B cells in peripheral blood and lymphoid tissues in the face of normal T-cell number and function. XLA is caused by mutations in BTK, encoding Bruton tyrosine kinase. Bruton tyrosine kinase deficiency results in arrested B-cell development in the bone marrow due to a severe block in their transition from CD19+ pro-B cells to the cytoplasmic μ-positive pre–B-cell stage. B cells of obligate heterozygote females are normal. However, the active X chromosome in the B cells is exclusively that which harbors the normal BTK allele, indicative of the critical role of Bruton tyrosine kinase in B-cell maturation.
Male infants with X-linked agammaglobulinemia (XLA) frequently become symptomatic late in the first year of life following the consumption of placentally acquired maternal immunoglobulins. However, affected males of the same family may vary in clinical disease onset and severity. Recurrent infections, especially with encapsulated pyogenic organisms, commonly involve the upper and lower respiratory tract, causing pneumonia, otitis, purulent sinusitis, and bronchiectasis. Additional infections include meningitis, sepsis, pyoderma, osteomyelitis, and giardiasis. Although most infections respond to appropriate antibiotics, the disease would eventually prove fatal unless immunoglobulin therapy is instituted. In its absence, many children with XLA develop progressive bronchiectasis and ultimately die of pulmonary or other end organ complications.
Children with XLA have an increased susceptibility to infection with enteroviral infections, including echo, coxsackie, and both wild-type and vaccine-associated polio virus. These agents can cause a dermatomyositis-like syndrome or fatal chronic encephalitis. The use of immunoglobulin replacement therapy has markedly decreased chronic enteroviral infection in patients with X-linked agammaglobulinemia, and high-dose therapy can be effective in therapy of acute infections. Chronic inflammation and swelling of the large joints, which resembles rheumatoid arthritis, develop in one third to one half of these children before the diagnosis is established, but resolves with the institution of immunoglobulin replacement.
Serum IgG levels are usually less than 1 g/L, and serum IgA and IgM levels are less than 1% of adult values. In most patients, the B cells are less than 0.2% of peripheral blood lymphocytes. The lymph nodes and tonsils are small and lack germinal centers and plasma cells. A positive family history of affected brothers or maternal uncles is supportive of the diagnosis. Carrier females can be identified by assessing X chromosome skewing in their B-cell population or by direct mutation analysis.
Immunoglobulin replacement therapy is a life-saving and effective treatment in X-linked agammaglobulinemia. Antibiotics should be used as appropriate for acute infections. Patients with X-linked agammaglobulinemia do not produce antibodies upon immunization, and they should not be given vaccines.
Patients with autosomal recessive agammaglobulinemia suffer from a heterogeneous set of defects affecting B-cell development. About one third have mutations in the μ-heavy chain gene. Others have defects in components of the pre–B-cell receptor, including the surrogate light chain (IGLL1 or λ5), the B-cell receptor–associated signal transducers CD79A (Iga) and CD79B (Igb), or the B-cell linker protein BLNK (see Fig. 188-2). In a few patients, the disease cause(s) remains unknown. Clinically, autosomal recessive agammaglobulinemia is very similar to X-linked agammaglobulinemia, although there is a tendency for patients with autosomal recessive agammaglobulinemia to present at a younger age and with more severe complications. The spectrum of infections is identical to that of X-linked agammaglobulinemia. Therapy mirrors that for X-linked agammaglobulinemia, including immunoglobulin replacement and antibiotics.
 HYPER-IgM SYNDROMES
HYPER-IgM SYNDROMES
B-cell intrinsic immunoglobulin class switch recombination (Ig-CSR) deficiencies, previously termed hyper-IgM syndromes, are genetically determined conditions characterized by normal or elevated serum IgM levels and an absence or very low levels of IgG, IgA, and IgE.37 The secondary antibody response is characterized by the production of immunoglobulins of various isotypes with high affinity for antigens by means of two distinct processes: class-switch recombination and somatic hypermutation. Class-switch recombination allows the replacement of the IgM heavy chain (Cμ) with heavy chains of different isotypes (Cg1-4 for IgG1-4, Ca for IgA, or Ce for IgE). This results in the production of immunoglobulins of different isotypes with distinct functional properties while leaving the immunoglobulin variable sequences (VDJ), and hence the antibody specificity and affinity, intact. Somatic hypermutation is the process through which mutations are introduced into the variable regions of immunoglobulins, resulting in a higher affinity and specificity for antigen. The hyper-IgM syndromes are typically associated with impaired class-switch recombination and, to a variable extent, defective somatic hypermutation.
X-linked hyper-IgM syndrome results from loss of function mutations in CD40L, encoding CD40 ligand.38 This is an inducible T-cell surface protein expressed on activated T helper cells that serves as a counter-receptor for the tumor necrosis factor family member CD40, which is expressed on B cells. Failure to engage CD40 by activated T cells blocks class-switch recombination and impairs somatic hypermutation (see Fig. 188-2). Consequently, patients with X-linked hyper-IgM syndrome have profoundly decreased serum concentrations of IgG and IgA levels in the face of normal or elevated IgM levels. They are particularly susceptible to infections with bacterial pathogens commonly associated with hypogammaglobulinemia. Because CD40 is also expressed on macrophages and dendritic cells where it mediates interactions with T helper cells, its deficiency is also associated with infections that are reflective of failure of those interactions. These infections include Pneumocystis jiroveci pneumonia, to which patients with X-linked hyper-IgM syndrome are particularly susceptible, and cryptosporidium-induced diarrhea and ascending cholangitis. Some patients with hypomorphic mutations also present with persistent anemia associated with parvovirus infection. Other manifestations include neutropenia and autoimmunity. An identical disease picture arises in both boys and girls due to rare autosomal recessive mutations in the CD40 gene.
Engagement of CD40 results in activation of the transcription factor nuclear factor-kappa B (NF-κB), which plays a critical role in class-switch recombination and somatic hypermutation. CD40 activates a kinase complex known as the inhibitors of NF-κB kinase, which mediates the phosphorylation of the inhibitors of NF-κB, leading to inhibitor of NF-κB degradation and NF-κB activation. Boys with X-linked hypohidrotic ectodermal dysplasia with immunodeficiency frequently present with a picture of hyper-IgM syndrome, with low levels of serum IgG and IgA, normal to increased IgM levels, and impaired antibody responses, particularly to polysaccharide antigens. In most cases, X-linked ectodermal dysplasia with immunodeficiency is caused by hypomorphic mutations in the zinc-finger domain of NF-κB essential modulator (also known as inhibitor of NF-κB kinase γ), a scaffolding protein that binds to the inhibitors of NF-κB kinase complex (NEMO).39 Hypomorphic mutations outside the zinc-finger domain of NF-κB essential modulator result in osteopetrosis, lymphedema, and atypical mycobacterial infections, but spare immunoglobulin production.
B-cell–restricted defects also result in hyper-IgM syndrome. Activation-induced cytidine deaminase, a DNA editing enzyme that is induced by T helper cell–B cell interaction, plays an essential role in both class-switch recombination and somatic hypermutation. Activation-induced cytidine deaminase deficiency results in an autosomal recessive form of hyper-IgM syndrome that presents with recurrent bacterial and viral infections of the respiratory, gastrointestinal, and central nervous systems. Lymphoid hyperplasia is a prominent feature of the disease. Though lacking the opportunistic infections associated with CD40L deficiency, they do suffer autoimmune manifestations, including arthritis and cytopenias. A phenotypically related though less common autosomal recessive hyper-IgM syndrome disorder results from deficiency in uracil N-glycosylase, which mediates deglycosylation and removal of deoxyuridine residues, a necessary step in the process of class-switch recombination.
 COMMON VARIABLE IMMUNODEFICIENCY
COMMON VARIABLE IMMUNODEFICIENCY
Common variable immunodeficiency syndrome represents a heterogeneous group of disorders characterized by the presence of hypogammaglobulinemia in the face of normal or low numbers of B cells in circulation.40-42 The hypogammaglobulinemia involves two or more immunoglobulin isotypes (IgG, IgA, and/or IgM), and impaired functional antibody responses include absent isohemagglutinins, poor responses to protein (diphtheria, tetanus) or polysaccharide vaccines (Streptococcus pneumoniae), or both. Additional clinical findings may include autoimmunity, granulomatous disease, and lymphoproliferative disease/neoplasia. Common variable immunodeficiency affects both sexes, and family members have a higher incidence of hypogammaglobulinemia, selective IgA deficiency, and autoimmune disease.
Most cases present during the second decade of life onward, although earlier onset is well established. Recurrent and chronic respiratory tract infections, particularly paranasal sinusitis, bronchitis, and pneumonia are prominent features. Gastrointestinal symptoms, including diarrhea, malabsorption, steatorrhea, and protein-losing enteropathy are common, resulting from complications of bacterial overgrowth, jejunal villous atrophy. Intestinal nodular lymphoid hyperplasia is often prominent. Giardia lamblia infection is common and appears to be responsible for many of the gastrointestinal complications seen in these patients.
Patients with common variable immunodeficiency frequently have associated hematologic disorders, including pernicious anemia, hemolytic anemia (including Coombs test–positive hemolytic anemia), anemia from folate or vitamin B12 malabsorption, leukopenia, and/or thrombocytopenia. Autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematosus, and idiopathic thrombocytopenic purpura are seen in patients with common variable immunodeficiency and their relatives at a much higher incidence than in the general population.43 Another distinguishing feature is the frequent occurrence of noncaseating granulomas of the lungs, spleen, liver, and skin. An infectious etiology for these lesions has not been identified, but steroids and anti-tumor necrosis factor therapy have been reported to be helpful in their treatment.
Three distinct genetic lesions have been identified in small subsets of common variable immunodeficiency patients. The most prevalent of these are heterozygous mutations in transmembrane activator and CAML interactor (TACI), a component of a network of tumor necrosis factor ligand and receptor superfamily members that play essential roles in B-cell differentiation and antibodies.44,45 These include the ligands B-cell activation factor of the tumor necrosis factor family, a proliferation-inducing ligand, and their three receptors: B-cell activation factor of the tumor necrosis factor family receptor, B-cell maturation antigen, and TACI. Failure of this system is associated with autoimmune disease, lymphoproliferation, and antibody deficiency. TACI mutations act in a dominant negative manner to impair isotype switching and antibody production. TACI mutations account for about 5% of all common variable immunodeficiency syndrome patients.
Rare mutations give rise to two other subtypes of common variable immunodeficiency. The inducible T-cell costimulator, expressed on activated T cells in B-cell follicles (so called follicular T helper cells), interacts with the inducible T-cell costimulator ligand, which is expressed on the surface of B cells to enable germinal center formation and class-switch recombination. Inducible T-cell costimulator deficiency results in deficiency of follicular T helper cells and decreased peripheral B cell numbers. It impairs terminal B-cell differentiation and leads to hypogammaglobulinemia. A second set of mutations lead to the deficiency of CD19, a B-cell–specific surface protein that plays an important role in B-cell activation and differentiation. The number of B cells in the periphery is normal, whereas the number of memory B cells is decreased.
 X-LINKED LYMPHOPROLIFERATIVE DISEASE
X-LINKED LYMPHOPROLIFERATIVE DISEASE
In the majority of patients with X-linked lymphoproliferative disease (known as XLP1), the underlying defect involves mutations in SH2D1A, encoding the protein SAP, signaling lymphocyte activation molecule (SLAM)–associated protein. SAP is a 128-amino acid peptide composed of an SH2 domain that associates with SLAM family proteins. SAP controls signal transduction via SLAMs, and failure to regulate SLAM signaling results in an exaggerated yet ineffective T-cell response to EBV infections (see Fig. 188-2).46 SAP deficiency results in the absence of a small subset of T cells known as natural killer T cells that is involved in immune regulation and antitumor responses. Natural killer T-cell deficiency contributes to the immune dysregulation in this syndrome. SAP deficiency also impairs the development of the differentiation of follicular T cells, leading to inability to support B-cell differentiation and profound humoral immunodeficiency.
Affected males suffer from severe, often fatal infections with Epstein-Bar virus (EBV) associated with fulminant hepatitis, B-cell lymphomas, agranulocytosis, aplastic anemia, or acquired hypogammaglobulinemia. These complications result from uncontrolled polyclonal T- and B-cell expansion triggered by the EBV infection. Approximately half of the affected individuals die of fatal infectious mononucleosis. Survivors suffer from hypogammaglobulinemia or agammaglobulinemia and malignant lymphomas. Most of the lymphomas are extranodal, Burkitt type, many involving the ileocecum.
A second subset of patients with X-linked lymphoproliferative disease (known as XLP2) suffer from mutations in BIRC4 gene, encoding the X-linked inhibitor of apoptosis protein (XIAP).47 XLP2 is phenotypically similar to XLP1, except for an early onset splenomegaly, which is the first clinical manifestation of the condition. XLP2 is also associated with natural killer T-cell deficiency. Management of both XLP1 and XLP2 involves immunoglobulin replacement therapy and vigilance for lymphoid malignancies. Bone marrow transplantation provides definitive therapy if attempted early in life, ideally before Epstein-Barr virus infection sets in.
 TRANSIENT HYPOGAMMAGLOBULINEMIA OF INFANCY
TRANSIENT HYPOGAMMAGLOBULINEMIA OF INFANCY
The normal full-term newborn has a serum IgG level that is the same or sometimes slightly higher than the mother’s, reflecting the active transport of maternal IgG across the placenta during the last trimester of pregnancy. Infants do not begin significant synthesis of IgG until 2 to 3 months of age. Catabolism of the maternal IgG (half-life of 25–30 days) precipitates a physiologic hypogammaglobulinemia between 4 and 6 months of age. In transient hypogammaglobulinemia of infancy, there is an abnormal prolongation and accentuation of the physiologic hypogammaglobulinemia resulting from a delay in the onset of immunoglobulin synthesis.48 Transient hypogammaglobulinemia of infancy affects both male and female infants, and in some cases shows a familial occurrence. It usually presents at around 6 months of age, with recurrent infections, most commonly otitis media and bronchitis. The numbers of T and B lymphocytes are normal, and patients are capable of mounting high-titer antibody responses to diphtheria and tetanus toxoid vaccination, even though the immunoglobulin concentrations are low. A transient defect in T helper-cell function has been implicated in transient hypogammaglobulinemia of infancy pathogenesis.
Patients with transient hypogammaglobulinemia of infancy usually recover spontaneously by the age of 2 to 4 years.49,50 The majority do not require immunoglobulin replacement therapy and can be followed conservatively with periodic determinations of serum immunoglobulin level. For those patients with severe recurrent infections or dangerously low immunoglobulin levels, replacement therapy is indicated; it is discontinued when there is evidence of normalized in vivo IgG production.
 SELECTIVE IgA AND IgG SUBCLASS DEFICIENCIES
SELECTIVE IgA AND IgG SUBCLASS DEFICIENCIES
IgA deficiency is the most prevalent primary immunodeficiency, with an estimated frequency those of European ancestry of 1 in 600.51 Primary IgA deficiency is caused by a defect of terminal lymphocyte differentiation, which leads to underproduction of serum and mucosal IgA; affected individuals have normal IgA genes. A number of non-immunoglobulin genes have been implicated in IgA deficiency. IgA in the serum is predominantly IgA1, and equal amounts of IgA1 and IgA2 are present in secretions. The overwhelming majority of IgA-deficient individuals are deficient in both subclasses.
Most IgA-deficient individuals are asymptomatic, but some suffer from recurrent respiratory and gastrointestinal tract infections. There is also an increased incidence of allergic and autoimmune diseases. Many develop anti-IgA antibodies and are at high risk for anaphylactoid reactions after receiving blood products, including immunoglobulin preparations, that contain IgA. A subset of those with IgA deficiency and recurrent infections also have deficiencies of IgG2 and IgG4 subclasses, indicating a more general abnormality in antibody production. The genetic cause of IgA deficiency remains unknown, although a strong association to genes within the MHC (HLA), in particular the class II and the class III regions, has been found.
There have been many diseases reported in association with IgA deficiency, particularly autoimmune diseases. The most common association is with coeliac disease (CD), which has special significance since CD is usually diagnosed by detection of specific IgA antibodies that are obviously lacking in IgA deficiency. There is no specific treatment for patients with symptomatic IgA deficiency.
Selective IgG subclass deficiency affects one or more of the IgG subclasses, including IgG1 (approximately 65% of serum IgG), IgG2 (20–25%), IgG3 (5–10%), and IgG4 (less than 5%).52 Each IgG subclass corresponds to a specific constant chain gene and is endowed with distinct effector functions. In most individuals with IgG subclass deficiency, the deficient subclass(es) are produced at low levels despite normal constant chain gene(s). Most individuals with IgG subclass deficiency are asymptomatic, but a few suffer from recurrent sinopulmonary infections and diarrheal illnesses with bacterial and viral pathogens. Symptomatic patients frequently have more than one affected IgG subclass, commonly IgG2 and IgG4, in isolation or together with IgA deficiency. A clue to the diagnosis is the presence of borderline or low-normal levels of IgG in the face of recurrent infections. Measurement of IgG subclass serum concentrations is diagnostic. Immunoglobulin replacement therapy is beneficial in symptomatic patients.53 Subclass deficiencies may normalize in some children with maturation.54
DISORDERS OF IMMUNE REGULATION
T regulatory cells (Treg) act to prevent autoimmunity and exuberant immune responses to infectious agents and allergens. Treg cells are programmed in the thymus and emerge as a distinct CD4+ T-cell lineage (see Fig. 188-1). They are dependent on the cytokine IL-2 for survival and function, and they constitutively express high levels of IL-2 receptor alpha chain (CD25). They also express high levels of the forkhead-family transcription factor forkhead box p3 (FOXP3), which is indispensable for Treg cell differentiation. FOXP3 deficiency precipitates a syndrome of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX).55-57 Affected males present in infancy with diarrhea; polyendocrinopathies, especially type I diabetes; and eczema, food allergies, and elevated serum IgE levels, reflecting a state of allergic dysregulation. IPEX is usually fatal, but bone marrow transplantation can be curative. Autosomal recessive IPEX-like diseases result from mutations in CD25 and, in a milder form, the IL-2 receptor-coupled transcription factor STAT5b.
Absolute and/or functional Treg cell deficiency also contributes to immune dysregulation associated with immunodeficiency diseases, including Omenn and Wiskott-Aldrich syndromes. Other immune defects also give rise to immune dysregulatory features. SAP deficiency, the underlying cause of XLP, leads to dysregulated cytotoxic T-cell responses to Epstein-Barr virus infections. Deficiency of components involved in cytotoxic T-lymphocyte granule exocytosis and effector functions, including perforin, RAB27, and MUNC-13-4, result in a syndrome of hemophagocytosis due to failure of cytotoxic T and NK cells to clear viral infections.
Two other disorders result from abnormalities in central and peripheral tolerance. Autoimmune polyendocrinopathy candidiasis, ectodermal dystrophy (Autoimmune polyendocrinopathy-type 1) is a rare monogenic autoimmune syndrome, which is caused by a defect in the AIRE gene on chromosome 21.57,58 AIRE controls the display of self antigens in the thymus, and its deficiency results in emergence of autoreactive T cells in the periphery. Patients develop autoimmunity with polyendocrinopathy, hepatitis, variable incidence of type I diabetes mellitus, and enteropathy, as well as a peculiar susceptibility to mucocutaneous candidiasis involving the oral cavity, esophagus, and nails. The FAS-FAS ligand system mediates cell suicide by activated T cells, a mechanism to downregulate the immune response. Mutations in FAS, its ligand, and caspase enzymes downstream from FAS lead to an autoimmune lymphoproliferative syndrome with autoimmunity and increased susceptibility to lymphoid malignancies.59,60
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


