Alterations
P53 signature
Serous tubal intraepithelial lesion (STIL)
Serous tubal intraepithelial carcinoma (STIC)
High-grade serous carcinoma
P53
Mutated (LOH)
Mutated
Mutated (LOH)
Mutated (LOH)
BRCA1/BRCA2
Mutated/LOH
Mutated/LOH/hypermethylated
Genes (FISH)
CCNE1 Amp
hTERT Amp
CCNE1 Amp
hTERT Amp
Protein expression
Pax8+, Bcl2+, Pax2−, γH2AX+
Up—CCNE1, p16, stathmin
Up—CCNE1, Rsf1, FASN, p16, stathmin, HMGA2, increase in CD68+ cells
Down—LKB1, FoxoA3, Rb1
Up—CCNE1, p16, stathmin, Rsf1, FASN
Down—LKB1, FoxoA3, Rb1
Chromosomal copy number alterations
Deleted—1p31.1, 1q21.1, 6p21.3, 8p11.2, 11q12.3, 12p13.3, 12q24.3, 15q11.2
Amplified—3q26.1, 4q13.2, 14q32.2, 20q13.2
Deleted—2p14, 2q31.1, 3q22.3, 3q26.3, 6p21.3, 6p11.2, 11q12.2-13.3, 18q12.1, 19p13.1, 20p13-p11.2, 22q13.3
Amplified—4p16.3, 8q13.1-q24.3, 10q26.3, 11q15.5-15.4, 12a12-13.1, 12q24.33, 12q24.4, 16q13.3 18 19q13.2-13.43
Extensive genomic rearrangements (TCGA 2011)
A consistent impediment to reducing mortality in HGSC has been the inability to diagnose HGSC at an early and potentially curable time in the disease course. Large trials using both transvaginal ultrasound and the serum marker CA125 failed to demonstrate an impact on mortality when using both modalities for ovarian cancer screening [2]. While new targeted therapies offer promise in the improved management of HGSC, it is possible that there will not be a major impact on HGSC mortality without more effective preventative and screening modalities. Consequently, there is a need to understand the molecular and genetic events which precede clinically evident HGSC. Until relatively recently, this was a serious challenge, in large part because there was no known histological precursor of HGSC. More knowledge about the classification and the natural history of the HGSC precursors, now recognized to exist in the fallopian tube, and their rate of progression to invasive carcinoma, is required before more effective early detection strategies can be developed [3, 4].
Historical Perspective
Classification of Serous Carcinoma
Until recently, the most common type of (surface) epithelial ovarian cancer was classified as serous carcinoma (also known previously as papillary serous carcinoma , papillary serous cystadenocarcinoma ) and graded using various three-tiered systems which were subjective and predicated loosely on architectural patterns and/or nuclear pleomorphism; the Silverberg–Shimizu grading system did apply objective criteria based on scoring of architecture, nuclear pleomorphism, and mitotic count. Despite the lack of consistency of grading, tumor grade was considered to be a prognostic factor in serous carcinoma.
Unlike endometrioid and mucinous carcinoma histotypes, most cases of serous carcinoma were not believed to arise in association with serous borderline tumors. In 1996, Kurman and his collaborators described a subset of serous borderline tumors that were at increased risk of recurrence and death [5]. The term noninvasive micropapillary carcinoma was proposed to separate these tumors from the usual type of serous borderline tumor and to link it to the invasive counterpart, a cytologically low-grade carcinoma which we now recognize as low-grade serous carcinoma (LGSC) . Subsequent molecular pathology studies identified an increased frequency of K-ras mutations in LGSC and its precursors, but not in the more conventional serous carcinoma, which we now diagnose as high-grade serous carcinoma [6]. In contrast, mutations of the tumor suppressor TP53 were identified in most HGSC, now known to be present in 98 % of HGSC, but were not seen in LGSC [7]. At about the same time, Malpica and Silva described a two-tier grading system based on cytological atypia and mitotic count which stratified ovarian carcinoma into two prognostic groups. They also noted the association of the low-grade tumors with borderline tumors [8]. In light of these findings, Kurman proposed that serous carcinoma be divided into two distinct entities with two different pathways of tumorigenesis . This was the first step in elucidating the pathogenesis of HGSC.
The dualistic model of ovarian cancer, developed by Kurman et al., proposed that Type I cancers, including endometrioid, mucinous, clear cell, and low-grade serous carcinomas, have a stepwise progression from benign to borderline and to carcinomas, are slow growing, are diagnosed at an early stage, and have a relatively indolent course. In contrast, the Type II tumors, of which high-grade serous carcinoma is the most frequent example, are aggressive, rapidly growing, and present at an advanced stage of spread, with poorly understood precursor lesions [9]. Morphologically, high-grade serous carcinoma has variable architecture, including the classic papillary pattern, often with bridging and fusion of papillae resulting in slit-like spaces, solid, pseudo-endometrioid glandular pattern, and transitional-like pattern. Multiple patterns may exist in any one tumor. All HGSC have high-grade nuclei and a high mitotic rate, usually with more than 25 mitoses per 10 high power fields. Molecularly, unifying features of HGSC include mutation of the tumor suppressor gene TP53, chromosome instability, and a high proliferation rate, frequently associated with alterations of the retinoblastoma pathway [7, 10, 11].
HGSC Cell of Origin
HGSC has no known morphologic precursor lesion in the ovary, and therefore, little was known of the early molecular/genetic events of serous carcinogenesis, a major obstacle in identifying markers of predisposition or of early stage ovarian cancer. Epidemiological studies of ovarian cancer risk factors, such as infertility, and protective factors, such as increased parity and oral contraceptive use, suggested that an increased number of lifetime ovulations may play a role in the development of ovarian carcinoma [12–15]. The importance of reproductive risk factors seemed to support the traditional theory of ovarian cancer origin, the incessant ovulation theory, first proposed by Fathalla in 1971 [16]. In the absence of native epithelium in the normal ovary, the ovarian surface epithelium (OSE) was the favored ovarian cancer cell of origin . OSEs are a modified mesothelial layer, express calretinin, are PAX8 negative, and are non-ciliated, with few immune cells associated with the monolayer [17]. It was suggested that with each ovulatory event, OSE damage and subsequent repair eventually led to acquisition of genetic abnormalities leading to carcinogenesis. This theory required that OSE undergo metaplasia to an epithelial cell type prior to the events of malignant transformation, often within ovarian cortical epithelial inclusion cysts [18]. In further support of the OSE cell of origin , there were reports that (1) atypia (reported as dysplasia or ovarian intraepithelial neoplasia) was present in surface epithelium adjacent to early stage ovarian carcinoma, (2) an increased number of cortical inclusion cysts were present in nonmalignant ovaries contralateral to ovarian cancer (histotype not specified), and (3) OSE adjacent to ovarian cancer had a higher incidence of metaplastic and hyperplastic changes [19–22]. For the most part, however, common histological changes associated with cancer precursors, such as cytological atypia, cellular stratification, and mitotic activity, have not been identified or of present are extremely rare in the ovary, indicating that if OSE was indeed the cell of origin of ovarian serous carcinoma, any precursor lesion must be very transient or the molecular progression of the disease is not associated with a morphological counterpart. Most of the publications describing early cancers or noninvasive cancer precursors within the ovary were reported prior to the routine histological examination of the fallopian tube. It is possible that the few images published purporting to represent dysplasia and early intraepithelial carcinoma within cortical inclusions actually represent cancerization of cortical cysts [23].
Convincing evidence in support of the fallopian tube epithelium (FTE) as the cell of origin emerged as pathologists began to examine the ovaries and fallopian tubes after risk-reducing surgery in women at high genetic risk of ovarian cancer. The two candidates for cell of origin, FTE and OSE , share common embryological origin, as well as close anatomic proximity. The Mullerian duct, which forms the fallopian tube, uterus, and endocervix, is formed by invagination of the embryonic coelomic epithelium, which also gives rise to mesothelium, including the OSE. The tube, lined by the hormonally sensitive FTE, has functions in ovum pickup and transport, facilitation of fertilization, and support of preimplantation embryo dev elopment in the first days after fertilization. It is divided into the interstitial portion (within the uterus), the isthmus, the ampulla, and the fimbria. Of particular note, and an important candidate for the source of serous carcinoma, is the FTE covering the fimbria, which has fingerlike projections in close contact to the OSE and ovarian surface and directly exposed to the peritoneal cavity (Fig. 1.1). It is of interest to note that genetic mouse models deleting BRCA1, Rb1, and TP53 genes from the OSE resulted in leiomyosarcomas [24] not high-grade serous carcinoma, and in contrast, targeted deletion of BRCA1, TP53, and PTEN in fallopian tube epithelia resulted in high-grade serous carcinoma with phenotypic and genomic alterations congruous with human HGSC [25].
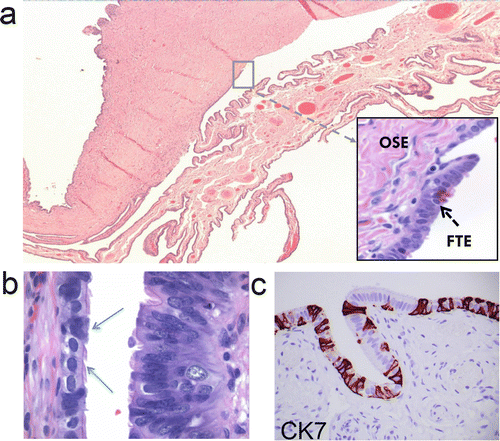
Fig. 1.1
Normal fallopian tube. (a) Single plica of the tubal fimbria is attached to the ovarian surface. The picture insert shows a higher magnification with a transition from tubal-type epithelium (FTE) to ovarian surface epithelium (OSE ) . (b) Normal tubal mucosa with varied stratification and a mixture of ciliated and secretory cells. (c) CK7 immunohistochemistry highlights the distribution of secretory cells (CK7 positive) and ciliated cells (CK7 negative)
BRCA and HGSC
A major advance in our understanding of HGSC tumorigenesis was the discovery in the mid-1990s that more than 90 % of hereditary ovarian cancers were associated with inherited germline mutations of BRCA1 and BRCA2. BRCA1/BRCA2 proteins have multiple functions including a critical role in the repair of double-stranded DNA breaks through homologous recombination. Loss of either BRCA1 or BRCA2 protein leads to deficient double-stranded DNA repair, which in turn increases the risk of chromosomal rearrangements. The lifetime risk of developing ovarian cancer is 40–60 % for BRCA1 mutation carriers and 10–20 % for BRCA2. Epidemiological studies indicated a predominance of HGSC in hereditary ovarian cancer, with a lack of mucinous carcinomas and borderline tumors [26, 27]. Blinded histopathological review using current definitions of histological classification demonstrates that the BRCA-deficient cancers are exclusively HGSC type [28–30].
The efficacy of risk-reducing salpingo-oophorectomy in women at high risk based on family history or germline mutations of BRCA1 or BRCA2 in preventing HGSC has been demonstrated, reducing the lifetime risk of HGSC to 5 % and decreasing mortality from all causes by 77 % [31, 32]. Because women with BRCA mutations are at the highest risk of developing HGSC, one might expect that if morphologic precursors of HGSC exist within the ovary, they would be detected in prophylactic oophorectomy specimens. However, a preliminary non-blinded report of histological features differentiating “cancer-prone” ovaries from ovaries at lower risk was not validated by other authors in more carefully controlled studies, nor was a reproducible histologic cancer precursor identified in the ovaries from high-risk patients [20, 33–38].
The addition of carcinoma of the fallopian tube to the list of BRCA1-/BRCA2-associated malignancies led to more comprehensive examination of the fallopian tube in prophylactic specimens. Support for the role of FTE in HGSC tumorigenesis was soon discovered, by the discovery of occult carcinomas and descriptions of putative cancer precursors in the fallopian tube epithelium [39–46].
Historically, precursor lesions of FTCa were not well defined, and the terms mucosal epithelial proliferation (MEP) , hyperplasia, and dysplasia have been applied variably in the literature to histological lesions of the FT epithelium (FTE). Mild hyperplasia of the FTE was reported to be a frequent finding and considered to be a normal, non-pathologic finding, but epithelial proliferation with nuclear atypia, which was described in moderate and severe MEP of FTE, was abnormal [47].
The finding of occult cancers and cancer precursors in the fallopian tubes of women at genetic high risk of serous carcinoma indicated an important and previously unsuspected role of the tubal epithelium (FTE) in BRCA mutation-associated serous carcinogenesis. These findings suggested an etiology of hereditary serous carcinoma other than that of malignant transformation of OSE cells. Early spread from a small clinically undetected carcinoma of the tubal fimbria, which is in direct contact with the peritoneal cavity and with the ovarian surface, would explain the lack of early detection by current technologies and the formation of ovarian masses, because the ovary is a fertile soil for growth of metastatic carcinomas from multiple sites. It would also explain the frequent peritoneal spread early in the disease course and many cases of presumed primary peritoneal carcinoma. While the observations leading to this hypothesis pertain to carriers of germline mutations, it seemed possible they would also apply to the more common sporadic serous carcinomas, which share molecular alterations with hereditary epithelial ovarian cancer, including loss of function of BRCA proteins [10, 48, 49].
High-Grade Serous Carcinoma, Occult
The rare finding of clinically incidental serous carcinoma was first reported in 1965 and was described further in a larger series by Bell and Scully in 1994[50]. It was noted in these early reports of “early de novo carcinoma” that despite the small size of the lesions, present within the ovarian cortex or on the ovarian surface and measuring up to 7 mm, an adverse outcome including recurrence and death was possible. The increase in prophylactic surgery in mutation carriers and the more comprehensive histological examination of salpingectomy specimens in recent years has led to the discovery of an increasing number of low-volume cancers at an early stage.
By definition , occult carcinoma is not detected preoperatively, and transvaginal ultrasound and serum CA125 are frequently reported as normal within the year prior to risk-reducing salpingo-oophorectomy (RRSO) . The first reports of occult carcinoma in BRCA mutation carriers indicated a range of 2.3–10.4 % incidence of occult carcinoma at the time of RRSO, and there was a surprisingly high incidence of carcinomas involving the distal end, fimbria, of the fallopian tubes [41, 45, 51, 52]. Ovarian involvement was also detected, but, in at least some of the cases, if the tube was carefully examined, the ovarian involvement was metastatic from the fallopian tube (Fig. 1.2). The frequency is higher in reports when examination of the fallopian tubes and ovaries is performed by meticulous fine sectioning and by consistent review limited to gynecologic pathologists at a single institution and lower in multi-institution studies without centralized pathology review. The incidence also varies with age of the patient at the time of RRSO and is more frequent with documented germline mutations of BRCA1/BRCA2 and more frequent with BRCA1 than BRCA2 mutations; it varies with the type of mutation (known deleterious, etc.) and, according to some studies, on whether the patient has a prior history of breast cancer [53, 54].
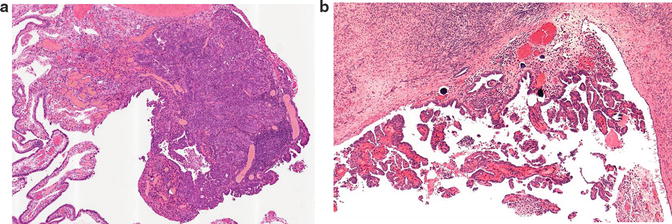
Fig. 1.2
Occult invasive carcinoma. (a) High-grade serous carcinoma in the tubal fimbria, measuring 1.6 mm, with adjacent serous tubal intraepithelial carcinoma. (b) Surface deposits of carcinoma on the ipsilateral ovary. The patient was a 44-year-old BRCA1 mutation carrier. Reprinted with permission [45]
Macroscopic
In the majority of cases, careful macroscopic examination will be unremarkable, but if visible, small pale nodules may be detected involving the fimbria, distal fallopian tube, or ovary, usually the ovarian surface. Most cases involve the fallopian tube or the fallopian tube and ovary, with a minority of cases involving only the ovary. Even though these tumors are not detected clinically, the stage of the carcinoma ranges from stage 1A to 3C [32, 51, 55, 56].
Microscopic
Occult carcinomas in RRSO specimens are usually HGSC [45, 54, 55]. Like clinically evident HGSC, architectural features vary, but they often have a mixed solid/papillary architecture. Because many of the carcinomas involve the distal end of the fallopian tube, there may also be deposits of tumor on the ovarian surf ace (Fig. 1.2).
Outcome
The carcinomas involving the fimbria, which are in close contact with the ovarian surface and are directly exposed to the peritoneal cavity, may be associated with microscopic spread to the ipsilateral ovary and the peritoneal cavity and have a significant risk of recurrence, reported to be as high as 43 %, despite the small size of the primary tumor [56, 57]. It is possible that undetected occult tubal carcinoma account for a significant proportion of cases diagnosed as primary peritoneal carcinoma in women with germline mutations. There have been few studies documenting the follow-up of patients presenting with occult, low-volume disease, but it appears that even with early stage, low-volume disease, and with adjuvant chemotherapy, there is still a significant risk of recurrence [57]. Peritoneal spread may be present with a distal tubal carcinoma of only a few millimeters maximum dimension [45].
Molecular Pathology
HGSC is a genetically unstable tumor, characterized by a varied histomorphology unified by marked pleomorphism, a high mitotic rate, and biomarker expression reflective of the most common molecular alterations. The latter includes the near-ubiquitous presence of a mutation in the tumor suppressor p53 ( TP53), resulting in either overaccumulation of p53 protein by immunohistochemistry (missense—60 % of analyzed cases) or complete loss of protein expression (frameshift/splicing junctions/nonsense—39 % of analyzed cases). Mutations of p53 are already present in early stage HGSC, and mutant TP53 is likely an essential driver mutation required for the early pathogenesis of HGSC. HGSC demonstrates widespread intratumoral heterogeneity in mutation, copy number, and expression profiles, indicating complex and highly individual evolutionary routes in HGSC progression. The only somatic mutation present within all samples, i.e., common in multiple tumor sites and in multiple tumor patients, was TP53 mutation; TP53 mutation, the most stable genomic feature of HGSC, appears to be the common route to malignant transformation [58].
Recently, Hunter and colleagues reported no difference in the level of genomic aberration observed in early low-volume occult tubal carcinomas compared with high-grade serous carcinomas, suggesting that, at least in BRCA1/BRCA2 mutation carriers, genomic instability is an early event in HGSC carcinogenesis [59].
Serous Tubal Intraepithelial Carcinoma
Intraepithelial lesions with morphological features of malignancy but no evidence of stromal invasion, now known as serous tubal intraepithelial carcinoma (STIC) , are detected in any one of the three clinical settings : (1) HGSC of presumed ovarian, tubal, peritoneal origins, (2) in prophylactic salpingectomy specimens from BRCA1/BRCA2 mutation carriers, and (3) rarely as an incidental finding in routine surgical specimens. These lesions have been recognized and reported in the past and were thought by some to represent evidence of multicentric tumorigenesis in Müllerian-type epithelium [60]. Other terms used in the literature include dysplasia, atypical mucosal epithelial proliferation, and carcinoma in situ [39, 47, 60].
Serous tubal intraepithelial carcinoma is defined as a localized lesion characterized by morphological atypia, abnormal p53 expression (reflecting the presence of a p53 mutation), and increased proliferation rate [18, 61]. Tubal intraepithelial carcinoma has been seen in association with HGSC for many years, but this finding was interpreted as evidence of a “field effect” of tumorigenesis —the secondary Müllerian system [60]. Careful examination of the fallopian tubes prophylactically resected from BRCA1/BRCA2 mutation carriers led to the description of STIC in the absence of invasive disease, an important finding which, along with the description of “dysplasia” in the RRSO specimens , supported the concept that HGSC, at least in BRCA mutation carriers, had its origin in the fallopian tube. Subsequently, STIC was reported in up to 61 % of tubes from patients with clinically evident HGSC, supporting the currently favored theory that STIC is the immediate precursor of HGSC in both hereditary and sporadic forms of HGSC [62–64].
Microscopic
Many STIC lesions are small, making the diagnosis sometimes challenging. The reproducibility of the diagnosis using morphological criteria alone is only moderate among experienced gynecological pathologists [65, 66]. Like HGSC, STIC has a variable histological appearance, and the morphological spectrum of changes is wide. STIC lesions have varying degrees of stratification, and some STIC has exfoliation of cells, sometimes with a growth pattern reminiscent of the slit-like spaces, epithelial “fractures” seen so commonly in the invasive counterpart (Fig. 1.3). Detachment of malignant-appearing cells may be associated with superficial implants of the ipsilateral ovary (Fig. 1.4).
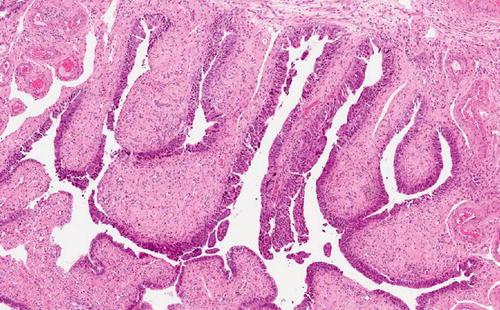
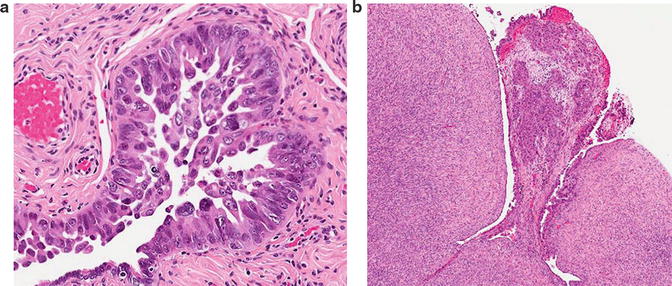
Fig. 1.3
Serous tubal intraepithelial lesion (STIC). A large lesion involving multiple plicae and demonstrating cellular stratification and detachment in addition to atypia and increased proliferation
Fig. 1.4
(a) Serous tubal intraepithelial lesion (STIC). This lesion has prominent tufting, with cell detachment. (b) A microscopic focus of high-grade serous carcinoma on the ipsilateral ovarian surface
Using current definitions which include the use of immunohistochemistry to improve diagnostic reproducibility, all cases of STIC have:
1.
Morphological atypia, which includes not necessarily all but a combination of the following features: nuclear enlargement, hyperchromasia, irregularly distributed chromatin, nucleolar prominence, loss of polarity, apoptosis, epithelial tufting, and mitotic activity
2.
Abnormal p53 expression by immunohistochemistry: diffuse intense nuclear positivity, or negativity in all lesional nuclei, the “null” pattern of expression
3.
Increased proliferation, with at least 10 % of tumor nuclei expressing Ki67 by immunohistochemistry
A diagnostic algorithm has been developed to improve the reproducibility of the diagnosis of tubal precursor lesions, independent of the pathologist’s level of experienc e (Fig. 1.5) [61, 66]. In brief, suspected tubal lesions are first assessed by morphological criteria and categorized as: (1) not suspicious for STIC, (2) suspicious for STIC, or (3) unequivocal for STIC. Immunostains help to further categorize the lesion.
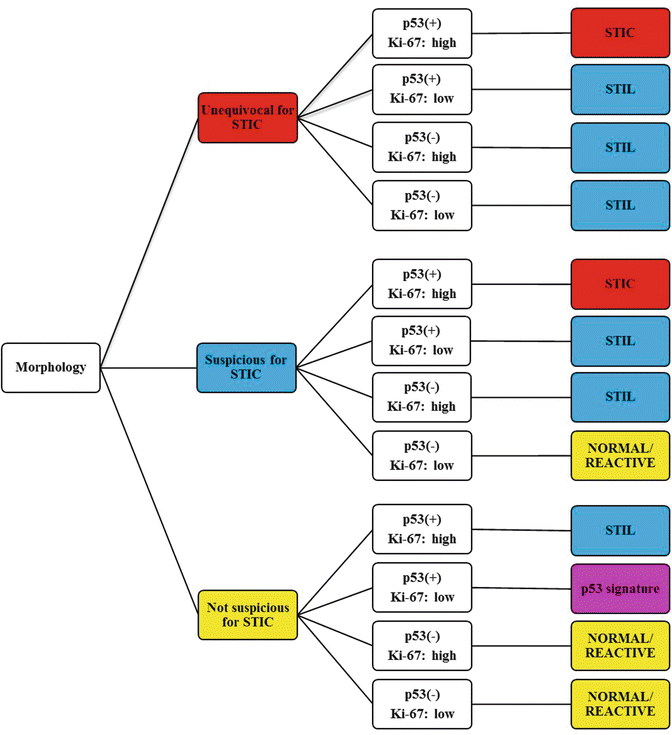
Fig. 1.5
Algorithm for the diagnosis of tubal intraepithelial lesions, using morphology and immunohistochemistry. Ki67 expression is considered high if positive in at least 10 % of lesion cells. P53 is considered positive with either a diffusely positive pattern or a null pattern of expression. Negative p53 in this chart reflects normal, wild-type expression. Reprinted with permission [61]
A diagnosis of STIC requires that a lesion is assessed:
1.
To be suspicious for STIC or unequivocal for STIC
2.
To have an abnormal p53 staining pattern, either intense nuclear positivity in greater than 75 % of the lesional cells or 0 % labeling (null pattern)
3.
To have increased proliferation as indicated by greater than 10 % of the lesional cells showing positive Ki67 staining
Lesions not meeting all of these criteria are not diagnosed as STIC, but may be diagnosed as serous tubal intraepithelial lesion (STIL), or p53 signature, or normal/reactive (Figs. 1.6 and 1.7).
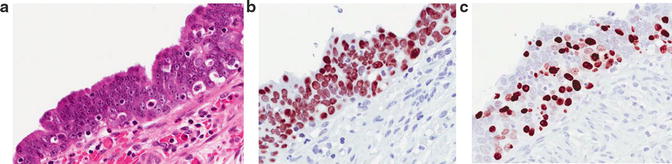
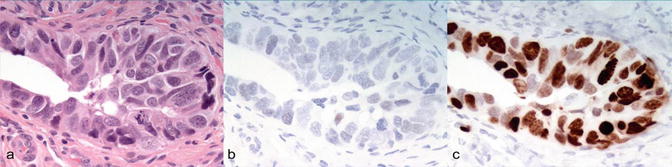
Fig. 1.6
Serous tubal intraepithelial lesion (STIC). (a) H&E . (b) P53 with diffuse nuclear overexpression . (c) Ki67
Fig. 1.7
Serous tubal intraepithelial lesion (STIC). (a) H&E. (b) P53 (null pattern of expression). (c) Ki67 [82]
An additional biomarker which has not yet been widely validated is laminin γ1, which has been proposed as an alternate biomarker of potential use in those STICs which have no p53 staining [67]. This marker may be useful in the diagnosis of STICs with a null pattern of p53 expression.
Molecular Pathology
STICs found in association with HGSC have been shown to have matching mutations of TP53 in 93 % of cases, supporting the clonal relationship between STIC and HGSC and providing further evidence that the STIC precedes HGSC [68]. The pattern of p53 expression by immunohistochemistry is highly concordant with the type of p53 mutation present [68]. Diffuse intense staining in STIC corresponds with missense mutations (overaccumulation of abnormal protein), and complete loss of staining corresponds to null mutations (due to splice, frameshift, and nonsense mutations). Weak, patchy, isolated cell positivity corresponds to wild-type TP53. Complete loss of staining is usually easily interpreted with wild-type positivity in the background uninvolved tubal epithelium.
Abnormal expression of p53 is present in close to 100 % of STICs, as in HGSC. Other translational changes, present in HGSC with varying frequencies, have also been detected by immunohistochemistry with similar levels of expression between STIC and synchronous HGSC: p16 overexpression (CDKN2A), loss of Retinoblastoma protein (Rb) [11], upregulation of the PI3K pathway (stathmin) [69], loss of FOXO3a [70], loss of Pax2 [70, 71], and loss of LKB1 [72]. Similarly, upregulation of oncogene products cyclin E, Rsf-1, and fatty acid synthase (FASN) is present in both STIC and HGSC [73]. In addition, shortened telomeres, important in early carcinogenesis, have been documented in STIC [74].
FISH studies have identified genomic aneuploidy in STIC lesions associated with HGSC, in chromosomes 1, 8, 11, and 17 [75]. Other studies have identified additional genomic similarities between STIC and HGSC, including overexpression of cyclin E [73] and amplification of human telomerase (hTERT) [74]. Additional genomic studies are ongoing, which will clarify the nature of the earliest genomic alterations further.
A concerted effort to molecularly annotate HGSC by the TCGA resulted in a seminal paper of a comprehensive catalog of the major genomic alterations within HGSC, including transcription, translation, and genomic rearrangements [10]. How early thes e changes occur in the disease course could be probed more comprehensively, and investigations are ongoing. However, STICs are often small and are diagnosed in formalin-fixed paraffin-embedded tissues, leading to technical challenges in the characterization of the genomic alterations which immediately precede HGSC. While much is still to be learned about the transcriptional, mutation, and genomic changes in STIC, it appears that STIC and invasive HGSC share many aberrations, indicating that although STIC is a noninvasive lesion, the cells have the propensity for metastasis without the requirement of invasion into adjacent stroma prior to peritoneal spread.
Outcome
The morphological features of STIC and the molecular associations reported to date suggest that STIC is a malignant lesion, and it has been recommended that STIC be staged as serous carcinoma, Stage 1A [76]. The clinical outcome of patients with STIC, in the absence of positive peritoneal washings or other diseases, is not yet well understood and cannot be predicted for an individual patient. There is at least one published report of a patient with a STIC but no evidence of invasive disease recurring with advanced-stage disease [57], although one small series indicates a favorable outcome [77]. Nevertheless, the accurate diagnosis of STIC clearly has significant clinical implications, although current information does not yet provide clear direction on how patients with a STIC diagnosis should be managed. Because STICs share molecular and genetic alterations with HGSC and at least 15 % of HGSCs are associated with germline mutations of BRCA1 or BRCA2, patients with an incidental diagnosis of STIC may also have a higher risk of carrying a deleterious mutation, and therefore, referral to a genetic counselor is likely indicated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


