 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Increased frequency of infections.
Increased frequency of infections.
 Ulceration of oral mucosa and gingivitis.
Ulceration of oral mucosa and gingivitis.
 Decreased absolute neutrophil count; normal numbers of red cells and platelets.
Decreased absolute neutrophil count; normal numbers of red cells and platelets.
 General Considerations
General Considerations
Neutropenia is an absolute neutrophil (granulocyte) count of less than 1500/μL in childhood, or less than 1100/μL between ages 1 month and 2 years. During the first few days of life, an absolute neutrophil count of less than 3500/μL may be considered neutropenia in term infants. Neutropenia results from absent or defective myeloid stem cells; ineffective or suppressed myeloid maturation; decreased production of hematopoietic cytokines (eg, granulocyte colony-stimulating factor [G-CSF] or granulocyte-macrophage colony-stimulating factor [GM-CSF]); decreased marrow release; increased neutrophil apoptosis; destruction or consumption; or, in pseudoneutropenia, from an increased neutrophil marginating pool (Table 30–5). A decrease in neutrophil mass diminishes delivery of these cells to areas where the balance favors bacterial proliferation and invasion.
Table 30–5. Classification of neutropenia of childhood.

The most severe types of congenital neutropenia include reticular dysgenesis (congenital aleukocytosis), Kostmann syndrome (severe neutropenia with maturation defect in the marrow progenitor cells), Shwachman syndrome (neutropenia with pancreatic insufficiency), neutropenia with immune deficiency states, cyclic neutropenia, and myelokathexis or dysgranulopoiesis. Genetic mutations for Chédiak-Higashi syndrome, Kostmann syndrome, Shwachman syndrome, and cyclic neutropenia and the newly described glucose-6-phosphatase catalytic subunit 3 (G6PC3) have been identified. Neutropenia may also be associated with storage and metabolic diseases and immunodeficiency states. The most common causes of acute neutropenia are viral infection or drugs, resulting in decreased neutrophil production in the marrow, increased peripheral turnover, or both. Severe bacterial infections may be associated with neutropenia. Although not commonly seen, neonatal alloimmune neutropenia can be severe and associated with infection. Autoimmune neutropenia occurs with chronic benign neutropenia of childhood, immunodeficiency syndromes, autoimmune disorders, or, in the newborn, as a result of passive transfer of antibody from the mother to the fetus. Malignancies, osteopetrosis, marrow failure syndromes, and hypersplenism usually are not associated with isolated neutropenia.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Acute severe bacterial or fungal infection is the most significant complication of neutropenia. Although the risk is increased when the absolute neutrophil count is less than 500/μL, the actual susceptibility is variable and depends on the cause of neutropenia, marrow reserves, and other factors. The most common types of infection include septicemia, cellulitis, skin abscesses, pneumonia, and perirectal abscesses. Sinusitis, aphthous ulcers, gingivitis, and periodontal disease also cause significant problems. In addition to local signs and symptoms, patients may have chills, fever, and malaise. In most cases, the spleen and liver are not enlarged. Staphylococcus aureus and gram-negative bacteria are the most common pathogens.
B. Laboratory Findings
Neutrophils are absent or markedly reduced in the peripheral blood smear. In most forms of neutropenia or agranulocytosis, the monocytes and lymphocytes are normal and the red cells and platelets are not affected. The bone marrow usually shows a normal erythroid series, with adequate megakaryocytes, but a marked reduction in the myeloid cells or a significant delay in maturation of this series may be noted. Total cellularity may be decreased.
In the evaluation of neutropenia (eg, persistent, intermittent, cyclic), attention should be paid to the duration and pattern of neutropenia, the types of infections and their frequency, and phenotypic abnormalities on physical examination. A careful family history and blood counts from the parents are useful. If an acquired cause, such as viral infection or drug, is not obvious as an acute cause and no other primary disease is present, WBC counts, white cell differential, and platelet and reticulocyte counts should be completed twice weekly for 6 weeks to determine the possibility of cyclic neutropenia. Bone marrow aspiration and biopsy are most important to characterize the morphologic features of myelopoiesis. Measuring the neutrophil counts in response to corticosteroid infusion may document the marrow reserves. Other tests that aid in the diagnosis include measurement of neutrophil antibodies, immunoglobulin levels, antinuclear antibodies, and lymphocyte phenotyping to detect immunodeficiency states. Tissue culture of bone marrow is important for defining the numbers of stem cells and progenitors committed to the myeloid series or the presence of inhibitory factors. Cytokine levels in plasma or mononuclear cells can be measured directly. Some neutropenia disorders have abnormal neutrophil function, but severe neutropenia may preclude collection of sufficient cells to complete assays. Recent studies have documented abnormalities in an antiapoptotic gene, HAX1, and the elastase gene, ELA2, in Kostmann syndrome and ELA2 mutations in cyclic neutropenia. A mutation for Shwachman syndrome has been described. Increased apoptosis in marrow precursors or circulating neutrophils has been described in several congenital or genetic disorders.
 Treatment
Treatment
Underlying disorders should be identified and treated or associated agents should be eliminated. Infections should be aggressively assessed and treated. Prophylactic antimicrobial therapy is not indicated for afebrile, asymptomatic patients, but may be considered in rare cases with recurrent infections. Recombinant G-CSF will increase neutrophil counts in most patients; GM-CSF may be considered, but is less extensively used. Patients may be started on 3–5 mcg/kg/d of G-CSF (filgrastim) given subcutaneously or intravenously once a day. Depending on the counts, the dose may be adjusted to keep the absolute neutrophil count below 10,000/μL. Some patients maintain adequate counts with G-CSF given every other day or three times a week. Treatment will decrease infectious complications but may have little effect on periodontal disease. However, not all patients with neutropenia syndromes require G-CSF (eg, chronic benign neutropenia of childhood). Patients with cyclic neutropenia may have a milder clinical course as they grow older. Immunizations should be given if the adaptive immune system is normal. Hematopoietic stem cell transplant may be considered for patients with severe complications, especially those with severe congenital neutropenia.
 Prognosis
Prognosis
The prognosis varies greatly with the cause and severity of the neutropenia. In severe cases with persistent agranulocytosis, the prognosis is poor in spite of antibiotic therapy; in mild or cyclic forms of neutropenia, symptoms may be minimal and the prognosis for normal life expectancy excellent. G-CSF has the potential to prolong life expectancy. Up to 50% of patients with Shwachman syndrome may develop aplastic anemia, myelodysplasia, or leukemia during their lifetime. Patients with Kostmann syndrome also have a potential for leukemia, as do patients with neutropenia associated with some immune disorders. Hematopoietic stem cell transplant may be the only curative therapy for some disorders.
Audrain M: Autoimmune neutropenia in children: analysis of 116 cases. Pediatr Alergy Immunol 2011:22;494 [PMID: 21771084].
Boztug K: Extended spectrum of human glucose-6-phosphatase catalytic subunit deficiency: novel genotypes and phenotype variability in congenital neutropenia. J Pediatr 2012;160:674 [PMID: 22050868].
Boztug K: Genetic etiologies of severe congenital neutropenia. Curr Opin Pediatr 2011;23:21 [PMID: 21206270].
Carlsson G: Hematopoietic stem cell transplantation in severe congenital neutropenia. Pediatr Blood Cancer 2011;56:444 [PMID: 21072829].
Fioredda F: Infectious complications in children with severe congenital, autoimmune or idiopathic neutropenia: a retrospective study form the Italian neutropenia registry. Pediatr Infect Dis J 2012 [Epub ahead of print] [PMID: 23249920].
NEUTROPHILIA
Neutrophilia is an increase in the absolute neutrophil count in the peripheral blood to greater than 7500–8500/μL for infants, children, and adults. To support the increased peripheral count, neutrophils may be mobilized from bone marrow storage or peripheral marginating pools. Neutrophilia occurs acutely in association with bacterial or viral infections, inflammatory diseases (eg, juvenile rheumatoid arthritis, inflammatory bowel disease, Kawasaki disease), surgical or functional asplenia, liver failure, diabetic ketoacidosis, azotemia, congenital disorders of neutrophil function (eg, chronic granulomatous disease, leukocyte adherence deficiency), and hemolysis. Drugs such as corticosteroids, lithium, and epinephrine increase the blood neutrophil count. Corticosteroids cause release of neutrophils from the marrow pool, inhibit egress from capillary beds, and postpone apoptotic cell death. Epinephrine causes release of the marginating pool. Acute neutrophilia has been reported after stress, such as from electric shock, trauma, burns, surgery, and emotional upset. Tumors involving the bone marrow, such as lymphomas, neuroblastomas, and rhabdomyosarcoma, may be associated with leukocytosis and the presence of immature myeloid cells in the peripheral blood. Infants with Down syndrome have defective regulation of proliferation and maturation of the myeloid series and may develop neutrophilia. At times this process may affect other cell lines and mimic myeloproliferative disorders or acute leukemia.
The neutrophilias must be distinguished from myeloproliferative disorders such as chronic myelogenous leukemia and juvenile chronic myelogenous leukemia. In general, abnormalities involving other cell lines, the appearance of immature cells on the blood smear, and the presence of hepatosplenomegaly are important differentiating characteristics.
DISORDERS OF NEUTROPHIL FUNCTION
Neutrophils play a key role in host defenses. Circulating in capillary beds, they adhere to the vascular endothelium adjacent to sites of infection and inflammation. Moving between endothelial cells, the neutrophil migrates toward the offending agent. Contact with a microbe that is properly opsonized with complement or antibodies triggers ingestion, a process in which cytoplasmic streaming results in the formation of pseudopods that fuse around the invader, encasing it in a phagosome. During the ingestion phase, the oxidase enzyme system assembles and is activated, taking oxygen from the surrounding medium and reducing it to form toxic oxygen metabolites critical to microbicidal activity. Concurrently, granules from the two main classes (azurophil and specific) fuse and release their contents into the phagolysosome. The concentration of toxic oxygen metabolites (eg, hydrogen peroxide, hypochlorous acid, hydroxyl radical) and other compounds (eg, proteases, cationic proteins, cathepsins, defensins) increases dramatically, resulting in the death and dissolution of the microbe. Complex physiologic and biochemical processes support and control these functions. Defects in any of these processes may lead to inadequate cell function and an increased risk of infection.
 Classification
Classification
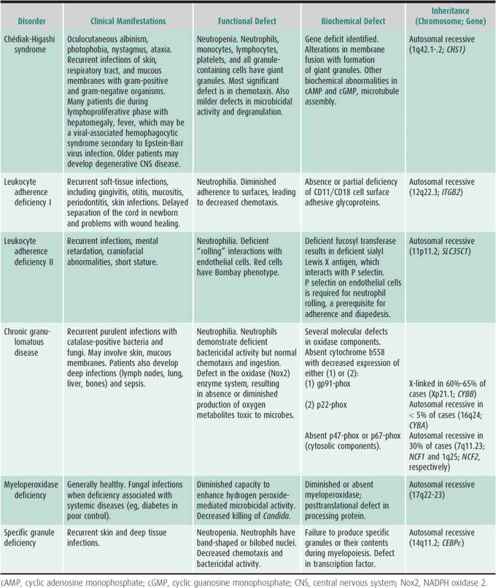
Table 30–6 summarizes congenital neutrophil function defects. Recently reported is variant CGD with p40phox deficiency manifested by inflammatory bowel disease. Also described is a syndrome of severe neutrophil dysfunction and severe infections associated with a mutation in a GTPase signaling molecule, Rac2. New syndromes of innate immune dysfunction include defects in interferon and interleukin (IL)-12 receptor and signaling pathways, leading to monocyte and macrophage dysfunction and defective toll-like receptor signaling pathways (IL-1 receptor-associated [IRAK] deficiency) associated with recurrent bacterial infections. Leukocyte adhesion deficiency (LAD) III is a disorder characterized by severe bleeding, impaired leukocyte adhesion, and endothelial inflammation, and is associated with mutations of FERMT3 gene, which encodes for a protein, Kindlin-3, critical for intracellular function of β integrins. Other congenital or acquired causes of mild to moderate neutrophil dysfunction include metabolic defects (eg, glycogen storage disease 1b, G6PC3 deficiency, diabetes mellitus, renal disease, and hypophosphatemia), viral infections, and certain drugs. Neutrophils from newborn infants have abnormal adherence, chemotaxis, and bactericidal activity. Cells from patients with thermal injury, trauma, and overwhelming infection have defects in cell motility and bactericidal activity similar to those seen in neonates.
 Clinical Findings
Clinical Findings
Recurrent bacterial or fungal infections are the hallmark of neutrophil dysfunction. Although patients will have infection-free periods, episodes of pneumonia, sinusitis, cellulitis, cutaneous and mucosal infections (including perianal or peritonsillar abscesses), and lymphadenitis are frequent. As with neutropenia, aphthous ulcers of mucous membranes, severe gingivitis, and periodontal disease are also major complications. In general, S aureus or gram-negative organisms are commonly isolated from infected sites; other organisms may be specifically associated with a defined neutrophil function defect. In some disorders, fungi account for an increasing number of infections. Deep or generalized infections, such as osteomyelitis, liver abscesses, sepsis, meningitis, and necrotic or gangrenous soft-tissue lesions, occur in specific syndromes (eg, leukocyte adherence deficiency or chronic granulomatous disease). Patients with severe neutrophil dysfunction may die in childhood from severe infections or associated complications. Table 30–6 summarizes pertinent laboratory findings.
 Treatment
Treatment
The mainstays of management of these disorders are anticipation of infections and aggressive attempts to identify the foci and the causative agents. Surgical procedures to achieve these goals may be both diagnostic and therapeutic. Broad-spectrum antibiotics covering the range of possible organisms should be initiated without delay, switching to specific antimicrobial agents when the microbiologic diagnosis is made. When infections are unresponsive or they recur, granulocyte transfusions may be helpful.
Chronic management may include prophylactic antibiotics. Trimethoprim-sulfamethoxazole and some other antibiotics (eg, rifampin) enhance the bactericidal activity of neutrophils from patients with chronic granulomatous disease. Some patients with Chédiak-Higashi syndrome improve clinically when given ascorbic acid. Recombinant γ-interferon decreases the number and severity of infections in patients with chronic granulomatous disease. Demonstration of this activity with one patient group raises the possibility that cytokines, growth factors, and other biologic response modifiers may be helpful in other conditions in preventing recurrent infections. Bone marrow transplant has been attempted in most major congenital neutrophil dysfunction syndromes, and reconstitution with normal cells and cell function has been documented. Combining genetic engineering with autologous bone marrow transplant may provide a future strategy for curing these disorders.
 Prognosis
Prognosis
For mild to moderate defects, anticipation and conservative medical management ensure a reasonable outlook. For severe defects, excessive morbidity and significant mortality still exist. In some diseases, the development of noninfectious complications, such as the lymphoproliferative phase of Chédiak-Higashi syndrome or inflammatory syndromes in chronic granulomatous disease, may influence prognosis.
Ambruso DR, Johnston RB Jr: Chronic granulomatous disease of childhood and common variable immunodeficiency. In: Chernick V, Boat TF (eds): Kendig’s Disorders of the Respiratory Tract in Children. Elsevier Saunders; 2012.
Dinauer MC: Disorders of neutrophil function: an overview. Methods Mol Biol 2007;412:489 [PMID: 18453130].
Kuhns DB: Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010;363:2600 [PMID: 21190454].
Robert P: A novel leukocyte deficiency III variant: kindlin-3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J Immunol 2011;186:5273 [PMID: 21441448].
Walach B: Lessons learned from phagocytic function studies in a large cohort of patients with recurring infections. J Clin Immunol 2012;32:454 [PMID: 222072252].
LYMPHOCYTOSIS
From the first week up to the fifth year of life, lymphocytes are the most numerous leukocytes in human blood. The ratio then reverses gradually to reach the adult pattern of neutrophil predominance. An absolute lymphocytosis in childhood is associated with acute or chronic viral infections, pertussis, syphilis, tuberculosis, and hyperthyroidism. Other noninfectious conditions, drugs, and hypersensitivity and serum sickness–like reactions cause lymphocytosis.
Fever, upper respiratory symptoms, gastrointestinal complaints, and rashes are clues in distinguishing infectious from noninfectious causes. The presence of enlarged liver, spleen, or lymph nodes is crucial to the differential diagnosis, which includes acute leukemia and lymphoma. Most cases of infectious mononucleosis are associated with hepatosplenomegaly or adenopathy. The absence of anemia and thrombocytopenia helps to differentiate these disorders. Evaluation of the morphology of lymphocytes on peripheral blood smear is crucial. Infectious causes, particularly infectious mononucleosis, are associated with atypical features in the lymphocytes, such as basophilic cytoplasm, vacuoles, finer and less-dense chromatin, and an indented nucleus. These features are distinct from the characteristic morphology associated with lymphoblastic leukemia. Lymphocytosis in childhood is most commonly associated with infections and resolves with recovery from the primary disease.
EOSINOPHILIA
Eosinophilia in infants and children is an absolute eosinophil count greater than 300/μL. Marrow eosinophil production is stimulated by the cytokine IL-5. Allergies, particularly those associated with asthma and eczema, are the most common primary causes of eosinophilia in children. Eosinophilia also occurs in drug reactions, with tumors (Hodgkin and non-Hodgkin lymphomas and brain tumors), and with immunodeficiency and histiocytosis syndromes. Increased eosinophil counts are a prominent feature of many invasive parasitic infections. Gastrointestinal disorders such as chronic hepatitis, ulcerative colitis, Crohn disease, and milk precipitin disease may be associated with eosinophilia. Increased blood eosinophil counts have been identified in several families without association with any specific illness. Rare causes of eosinophilia include the hypereosinophilic syndrome, characterized by counts greater than 1500/μL and organ involvement and damage (hepatosplenomegaly, cardiomyopathy, pulmonary fibrosis, and central nervous system injury). This is a disorder of middle-aged adults and is rare in children. Eosinophilic leukemia has been described, but its existence as a distinct entity is very rare.
Eosinophils are sometimes the last type of mature myeloid cell to disappear after marrow ablative chemotherapy. Increased eosinophil counts are associated with graft-versus-host disease after bone marrow transplant, and elevations are sometimes documented during rejection episodes in patients who have solid organ grafts.
BLEEDING DISORDERS
Bleeding disorders may occur as a result of (1) quantitative or qualitative abnormalities of platelets, (2) quantitative or qualitative abnormalities in plasma procoagulant factors, (3) vascular abnormalities, or (4) accelerated fibrinolysis. The coagulation cascade and fibrinolytic system are shown in Figures 30–4 and 30–5.
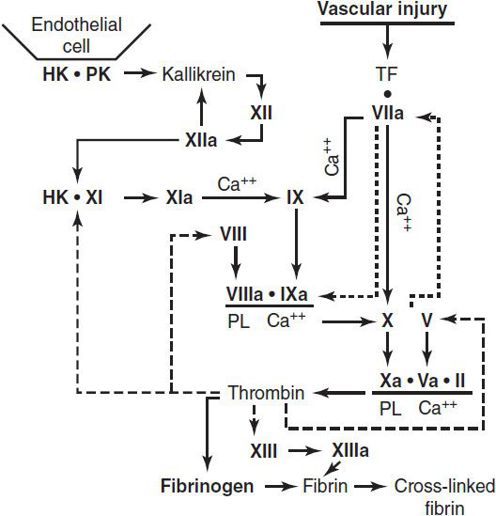
 Figure 30–4. The procoagulant system and formation of a fibrin clot. Vascular injury initiates the coagulation process by exposure of tissue factor (TF); the dashed lines indicate thrombin actions in addition to clotting of fibrinogen. The dotted lines associated with VIIa indicate the feedback activation of the VII-TF complex by Xa and IXa. Ca++, calcium; HK, high-molecular-weight kininogen; PL, phospholipid; PK, prekallikrein. (Reproduced, with permission, from Good-night SH, Hathaway WE [eds]: Disorders of Hemostasis & Thrombosis: A Clinical Guide, 2nd ed. McGraw-Hill; 2001.)
Figure 30–4. The procoagulant system and formation of a fibrin clot. Vascular injury initiates the coagulation process by exposure of tissue factor (TF); the dashed lines indicate thrombin actions in addition to clotting of fibrinogen. The dotted lines associated with VIIa indicate the feedback activation of the VII-TF complex by Xa and IXa. Ca++, calcium; HK, high-molecular-weight kininogen; PL, phospholipid; PK, prekallikrein. (Reproduced, with permission, from Good-night SH, Hathaway WE [eds]: Disorders of Hemostasis & Thrombosis: A Clinical Guide, 2nd ed. McGraw-Hill; 2001.)
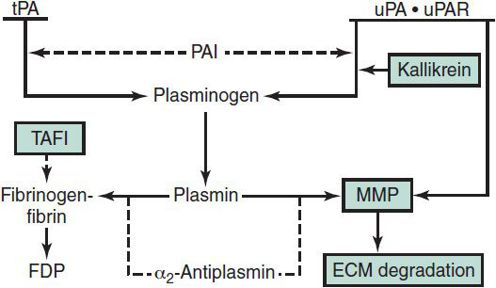
 Figure 30–5. The fibrinolytic system. Solid arrows indicate activation; dashed line arrows indicate inhibition. ECM, extracellular matrix; FDP, fibrinogen-fibrin degradation products; MMP, matrix metalloproteinases; PAI, plasminogen activator inhibitor; TAFI, thrombin activatable fibrinolysis inhibitor; tPA, tissue plasminogen activator; uPA, urokinase; uPAR, cellular urokinase receptor. (Reproduced, with permission, from Goodnight SH, Hathaway WE [eds]: Disorders of Hemostasis & Thrombosis: A Clinical Guide, 2nd ed. McGraw-Hill; 2001.)
Figure 30–5. The fibrinolytic system. Solid arrows indicate activation; dashed line arrows indicate inhibition. ECM, extracellular matrix; FDP, fibrinogen-fibrin degradation products; MMP, matrix metalloproteinases; PAI, plasminogen activator inhibitor; TAFI, thrombin activatable fibrinolysis inhibitor; tPA, tissue plasminogen activator; uPA, urokinase; uPAR, cellular urokinase receptor. (Reproduced, with permission, from Goodnight SH, Hathaway WE [eds]: Disorders of Hemostasis & Thrombosis: A Clinical Guide, 2nd ed. McGraw-Hill; 2001.)
The most critical aspect in evaluating the bleeding patient is obtaining detailed personal and family bleeding histories, including bleeding complications associated with dental interventions, surgeries, suture placement and removal, and trauma. Excessive mucosal bleeding is suggestive of a platelet disorder, von Willebrand disease (vWD), dysfibrinogenemia, or vasculitis. Bleeding into muscles and joints may be associated with a plasma procoagulant factor abnormality. In either scenario, the abnormality may be congenital or acquired. A thorough physical examination should be performed with special attention to the skin, oro- and nasopharynx, liver, spleen, and joints. Screening and diagnostic evaluation in patients with suspected bleeding disorders should initially include the following laboratory testing:
1. Prothrombin time (PT) to assess clotting function of factors VII, X, V, II, and fibrinogen.
2. Activated partial thromboplastin time (aPTT) to assess clotting function of high-molecular-weight kininogen, prekallikrein, XII, XI, IX, VIII, X, V, II, and fibrinogen.
3. Platelet count and size determined as part of a CBC.
4. Platelet functional assessment by platelet function analyzer-100 (PFA-100), template bleeding time, or whole blood platelet aggregometry.
5. Fibrinogen functional level by clotting assay.
The following laboratory tests may also be useful:
1. Thrombin time to measure the generation of fibrin from fibrinogen following conversion of prothrombin to thrombin, as well as the antithrombin effects of fibrin-split products and heparin. The thrombin time may be prolonged in the setting of a normal fibrinogen concentration if the fibrinogen is dysfunctional (ie, dysfibrinogenemia).
2. Euglobulin lysis time (ELT), if available, to evaluate for accelerated fibrinolysis if the preceding workup is nonrevealing despite documented history of pathologic bleeding. If the ELT is shortened, assessment of plasminogen activator inhibitor-1 and α2-antiplasmin is warranted, as congenital deficiency in these fibrinolytic inhibitors may cause hyperfibrinolysis. In ill patients, measurement of fibrin degradation products may assist in the diagnosis of DIC.
Goodnight SH, Hathaway WE (eds): Disorders of Hemostasis & Thrombosis: A Clinical Guide, 2nd ed. McGraw-Hill, 2001:41–51.
ABNORMALITIES OF PLATELET NUMBER OR FUNCTION
Thrombocytopenia in the pediatric age range is often immune-mediated (eg, ITP, neonatal auto- or alloimmune thrombocytopenia, heparin-induced thrombocytopenia), but is also caused by consumptive coagulopathy (eg, DIC, Kasabach-Merritt syndrome), acute leukemias, rare disorders such as Wiskott-Aldrich syndrome and type 2b vWD, and artifactually in automated cytometers (eg, Bernard-Soulier syndrome), where giant forms may not be enumerated as platelets by automated cell counters.
1. Idiopathic Thrombocytopenic Purpura
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Otherwise healthy child.
Otherwise healthy child.
 Decreased platelet count.
Decreased platelet count.
 Petechiae, ecchymoses.
Petechiae, ecchymoses.
 General Considerations
General Considerations
Acute idiopathic thrombocytopenic purpura (ITP) is the most common bleeding disorder of childhood. It occurs most frequently in children aged 2–5 years and often follows infection with viruses, such as rubella, varicella, measles, parvovirus, influenza, EBV, or acute and chronic HIV. Most patients recover spontaneously within a few months. Chronic ITP (> 12 months’ duration) occurs in 10%–20% of affected patients. The thrombocytopenia results from clearance of circulating IgM- or IgG-coated platelets by the reticuloendothelial system. The spleen plays a predominant role in the disease by forming the platelet cross-reactive antibodies and sequestering the antibody-bound platelets.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Onset of ITP is usually acute, with the appearance of multiple petechiae and ecchymoses. Epistaxis is also common at presentation. No other physical findings are usually present. Rarely, concurrent infection with EBV or CMV may cause hepatosplenomegaly or lymphadenopathy, simulating acute leukemia.
B. Laboratory Findings
1. Blood—The platelet count is markedly reduced (usually < 50,000/μL and often < 10,000/μL), and platelets frequently are of larger size on peripheral blood smear, suggesting accelerated production of new platelets. The white blood count and differential are normal, and the hemoglobin concentration is preserved unless hemorrhage has been significant.
2. Bone marrow—The number of megakaryocytes is increased. Erythroid and myeloid cellularity is normal.
3. Other laboratory tests—Platelet-associated IgG or IgM, or both, may be demonstrated on the patient’s platelets or in the serum. PT and aPTT are normal.
 Differential Diagnosis
Differential Diagnosis
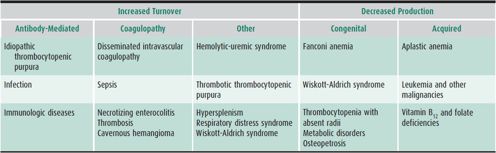
Table 30–7 lists common causes of thrombocytopenia. ITP is a diagnosis of exclusion. Family history or the finding of predominantly giant platelets on the peripheral blood smear is helpful in separating from thrombocytopenia that is hereditary. Bone marrow examination should be performed if the history is atypical (ie, the child is not otherwise healthy, or if there is a family history of bleeding), if abnormalities other than purpura and petechiae are present on physical examination, or if other cell lines are affected on the CBC. The importance of performing a bone marrow examination prior to using corticosteroids in the treatment for ITP remains controversial.
Table 30–7. Common causes of thrombocytopenia.
 Complications
Complications
Severe hemorrhage and bleeding into vital organs are the feared complications of ITP. Intracranial hemorrhage is the most serious (but rarely seen) complication, occurring in less than 1% of affected children. The most important risk factors for hemorrhage are a platelet count less than 10,000/μL and mean platelet volume less than 8 fL.
 Treatment
Treatment
A. General Measures
Treatment is optional in most children in the absence of bleeding. Aspirin and other medications (eg, NSAIDs such as Advil, Naproxin, etc) that compromise platelet function should be avoided. Bleeding precautions (eg, restriction from physical contact activities and use of helmets) should be observed. Platelet transfusion should be avoided except in circumstances of life-threatening bleeding, in which case emergent splenectomy may be considered. In this setting, administration of corticosteroids and IVIG is also advisable.
B. Corticosteroids
Patients with clinically significant but non–life-threatening bleeding (ie, epistaxis, hematuria, and hematochezia) and those with a platelet count of less than 10,000/μL may benefit from prednisone at 1–2 mg/kg/d for 2–3 weeks with a maximum dose of 60–80 mg/d. A higher dose initially (3–5 mg/kg/d) for 3–7 days may lead to faster count recovery. The dosage is then tapered and stopped. No further prednisone is given regardless of the platelet count unless significant bleeding recurs, at which time prednisone is administered in the smallest dose that achieves resolution of bleeding episodes (usually 2.5–5 mg twice daily). Follow-up continues until the steroid can again be discontinued, spontaneous remission occurs, or other therapeutic measures are instituted. Toxicity (Cushingoid facies, weight gain, change in behavior, hyperglycemia, and hypertension) is usually mild for short treatment courses.
C. Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) is the treatment of choice for severe, acute bleeding, and may also be used as an alternative or adjunct to corticosteroid treatment in both acute and chronic ITP of childhood. IVIG may be effective even when the patient is resistant to corticosteroids; responses are prompt and may last for several weeks. Most patients receive 0.8–1 g/kg/d for 1–2 days. Infusion time is typically 4–6 hours. Platelets may be given simultaneously during life-threatening hemorrhage but are rapidly destroyed. Adverse effects of IVIG are common, including transient neurologic complications in one-third of patients (eg, headache, nausea, and aseptic meningitis). These symptoms may mimic those of intracranial hemorrhage and necessitate radiologic evaluation of the brain. A transient decrease in neutrophil number may also be seen.
D. Anti-Rh(D) Immunoglobulin
This polyclonal immunoglobulin binds to the D antigen on RBCs. The splenic clearance of anti-D–coated red cells interferes with removal of antibody-coated platelets, resulting in improvement in thrombocytopenia. This approach is effective only in Rh(+) patients with a functional spleen. The time required for platelet increase is slightly longer than with IVIG. However, approximately 80% of Rh(+) children with acute or chronic ITP respond well. Significant hemolysis may occur transiently with an average hemoglobin concentration decrease of 0.8 g/dL. However, severe hemolysis occurs in 5% of treated children, and clinical and laboratory evaluation following administration is warranted in all patients. Rh(D) immunoglobulin is less expensive and infused more rapidly than IVIG but is more expensive than corticosteroids.
E. Splenectomy
Many children with chronic ITP have platelet counts greater than 30,000/μL. Up to 70% of such children spontaneously recover with a platelet count greater than 100,000/μL within 1 year. For the remainder, corticosteroids, IVIG, and anti-D immunoglobulin are typically effective treatment for acute bleeding. Splenectomy produces a response in 70%–80%, but it should be considered only after persistence of significant thrombocytopenia for at least 1 year. Preoperative treatment with corticosteroids, IVIG, or anti-D immunoglobulin is usually indicated. Postoperatively, the platelet count may rise to 1 million/μL. This reactive thrombocytosis is not associated with thrombotic complications in children. The risk of overwhelming infection (predominantly with encapsulated organisms) is increased after splenectomy, particularly in the young child. Therefore, the procedure should be postponed, if possible, until age 5 years. Administration of pneumococcal, H influenzae type b and meningococcal vaccines at least 2 weeks prior to splenectomy is recommended. Daily penicillin prophylaxis should be started postoperatively and continued at least until 5 years of age.
F. Rituximab (Anti-CD20 Monoclonal Antibody)
There have been no randomized trials for rituximab in children. The efficacy of treating childhood chronic ITP in several series and case studies has demonstrated a response rate of 60%. Because of significant adverse events, this therapy may be reserved for refractory cases with significant bleeding or as an alternative to splenectomy.
G. New Agents
One randomized clinical trial in children has been conducted with romiplostim, a thrombopoietin receptor agonist, with an 88% response rate and improved quality of life. Further studies in larger numbers of pediatric patients are needed to address the possibility that some patients with chronic ITP have a response in platelet production that is not maximally increased.
 Prognosis
Prognosis
Ninety percent of children with ITP will have a spontaneous remission. Features associated with the development of chronic ITP include female gender, age greater than 10 years at presentation, insidious onset of bruising, and the presence of other autoantibodies. Older child- and adolescent-onset ITP is associated with an increased risk of chronic autoimmune diseases or immunodeficiency states. Appropriate screening by history and laboratory studies (eg, antinuclear antibody) is warranted.
Blanchette V: Childhood immune thrombocytopenic purpura: diagnosis and management. Pediatr Clin North Am 2008;55:393 [PMID: 18381093].
Journeycake J: Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444 [PMID: 23233617].
Neunert C: The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190 [PMID: 21325604].
2. Thrombocytopenia in the Newborn
Thrombocytopenia is one of the most common causes of neonatal hemorrhage and should be considered in any newborn with petechiae, purpura, or other significant bleeding. Defined as a platelet count less than 150,000/μL, thrombocytopenia occurs in approximately 0.9% of unselected neonates. Several specific entities may be responsible (see Table 30–7); however, half of such neonates have alloimmune thrombocytopenia. Infection and DIC are the most common causes of thrombocytopenia in ill full-term newborns and in preterm newborns. In the healthy neonate, antibody-mediated thrombocytopenia (alloimmune or maternal autoimmune), viral syndromes, hyperviscosity, and major-vessel thrombosis are frequent causes of thrombocytopenia. Management is directed toward the underlying etiology.
A. Thrombocytopenia Associated With Platelet Alloantibodies (Neonatal Alloimmune Thrombocytopenia)
Platelet alloimmunization occurs in 1 out of approximately 350 pregnancies. Unlike in Rh incompatibility, 30%–40% of affected neonates are first-born. Thrombocytopenia is progressive over the course of gestation and worse with each subsequent pregnancy. Alloimmunization occurs when a platelet antigen of the infant differs from that of the mother, and the mother is sensitized by fetal platelets that cross the placenta into the maternal circulation. In Caucasians, alloimmune thrombocytopenia is most often due to human platelet antigen (HPA)-1a incompatibility. Sensitization of a mother homozygous for HPA-1b to paternally acquired fetal HPA-1a antigen results in severe fetal thrombocytopenia in 1 in 1200 fetuses. Only 1 in 20 HPA-1a–positive fetuses of HPA-1a–negative mothers develop alloimmunization. Other platelet-specific alloantigens may be etiologic. The presence of antenatal maternal platelet antibodies on more than one occasion and their persistence into the third trimester is predictive of severe neonatal thrombocytopenia; a weak or undetectable antibody does not exclude thrombocytopenia. Severe intracranial hemorrhage occurs in 10%–30% of affected neonates as early as 20 weeks’ gestation. Petechiae or other bleeding manifestations are usually present shortly after birth. The disease is self-limited, and the platelet count normalizes within 4 weeks.
If alloimmunization is associated with clinically significant bleeding, transfusion of platelet concentrates harvested from the mother is more effective than random donor platelets in increasing the platelet count. Transfusion with HPA-matched platelets from unrelated donors or treatment with IVIG or methylprednisolone to acutely block macrophage uptake of sensitized cells has also been successful in raising the platelet count and achieving hemostasis. If thrombocytopenia is not severe and bleeding is absent, observation alone is often appropriate.
Intracranial hemorrhage in a previous child secondary to alloimmune thrombocytopenia is the strongest risk factor for severe fetal thrombocytopenia and hemorrhage in a subsequent pregnancy. Amniocentesis or chorionic villus sampling to obtain fetal DNA for platelet antigen typing is sometimes performed if the father is heterozygous for HPA-1a. If alloimmunization has occurred with a previous pregnancy, irrespective of history of intracranial hemorrhage, screening cranial ultrasound for hemorrhage should begin at 20 weeks’ gestation and be repeated regularly. In addition, cordocentesis should be performed at approximately 20 weeks’ gestation, with prophylactic transfusion of irradiated, leukoreduced, maternal platelet concentrates. If the fetal platelet count is less than 100,000/μL, the mother should be treated with weekly IVIG. Delivery by elective cesarean section is recommended if the fetal platelet count is less than 50,000/μL, to minimize the risk of intracranial hemorrhage associated with birth trauma.
B. Thrombocytopenia Associated With ITP in the Mother (Neonatal Autoimmune Thrombocytopenia)
Infants born to mothers with idiopathic thrombocytopenic purpura (ITP) or other autoimmune diseases (eg, antiphospholipid antibody syndrome or systemic lupus erythematosus) may develop thrombocytopenia as a result of transfer of antiplatelet IgG from the mother to the infant. Unfortunately, maternal and fetal platelet counts and maternal antiplatelet antibody levels are unreliable predictors of bleeding risk. Antenatal corticosteroid administration to the mother is generally instituted once maternal platelet count falls below 50,000/μL, with or without a concomitant course of IVIG.
Most neonates with autoimmune thrombocytopenia do not develop clinically significant bleeding, and thus treatment for thrombocytopenia is not often required. The risk of intracranial hemorrhage is 0.2%–2%. If diffuse petechiae or minor bleeding are evident, a 1- to 2-week course of oral prednisone, 2 mg/kg/d, may be helpful. If the platelet count remains consistently less than 20,000/μL or if severe hemorrhage develops, IVIG should be given (0.8–1 g/kg daily for 1–2 days). Platelet transfusions are only indicated for life-threatening bleeding, and may only be effective after removal of antibody by exchange transfusion. The platelet nadir is typically between the fourth and sixth day of life and improves significantly by 1 month; full recovery may take 2–4 months. Platelet recovery may be delayed in breast-fed infants because of transfer of IgG to the milk.
C. Neonatal Thrombocytopenia Associated With Infections
Thrombocytopenia is commonly associated with severe generalized infections during the newborn period. Between 50% and 75% of neonates with bacterial sepsis are thrombocytopenic. Intrauterine infections such as rubella, syphilis, toxoplasmosis, CMV, herpes simplex (acquired intra- or postpartum), enteroviruses, and parvovirus are often associated with thrombocytopenia. In addition to specific treatment for the underlying disease, platelet transfusions may be indicated in severe cases.
D. Thrombocytopenia Associated With Kaposiform Hemangioendotheliomas (Kasabach-Merritt Syndrome)
A rare but important cause of thrombocytopenia in the newborn is kaposiform hemangioendotheliomas, a benign neoplasm with histopathology distinct from that of classic infantile hemangiomas. Intense platelet sequestration in the lesion results in peripheral thrombocytopenia and may rarely be associated with a DIC-like picture and hemolytic anemia. The bone marrow typically shows megakaryocytic hyperplasia in response to the thrombocytopenia. Corticosteroids, α-interferon, and vincristine are all useful for reducing the size of the lesion and are indicated if significant coagulopathy is present, the lesion compresses a vital structure, or the lesion is cosmetically unacceptable. If consumptive coagulopathy is present, heparin or aminocaproic acid may be useful. Depending on the site, embolization may be an option. Surgery is often avoided because of the high risk of hemorrhage.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


