 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Progressive pancytopenia.
Progressive pancytopenia.
 Macrocytosis.
Macrocytosis.
 Multiple congenital anomalies.
Multiple congenital anomalies.
 Increased chromosome breakage in peripheral blood lymphocytes.
Increased chromosome breakage in peripheral blood lymphocytes.
 General Considerations
General Considerations
Fanconi anemia, which is the number 1 inherited bone marrow failure syndrome, is characterized by defective DNA repair that is caused by a variety of genetic mutations. Inheritance is autosomal recessive, and this disease occurs in all ethnic groups; 75%–90% of affected individuals develop bone marrow failure in the first 10 years of life.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Symptoms are determined principally by the degree of hematologic abnormality. Thrombocytopenia may cause purpura, petechiae, and bleeding; neutropenia may cause severe or recurrent infections; and anemia may cause weakness, fatigue, and pallor. Congenital anomalies are present in at least 50% of patients. The most common include abnormal pigmentation of the skin (generalized hyperpigmentation, café au lait or hypopigmented spots), short stature with delicate features, and skeletal malformations (hypoplasia, anomalies, or absence of the thumb and radius). More subtle anomalies are hypoplasia of the thenar eminence or a weak or absent radial pulse. Associated renal anomalies include aplasia, horseshoe kidney, and duplication of the collecting system. Other anomalies are microcephaly, microphthalmia, strabismus, ear anomalies, and hypogenitalism.
B. Laboratory Findings
Thrombocytopenia or leukopenia typically occurs first, followed over the course of months to years by anemia and progression to severe aplastic anemia. Macrocytosis is virtually always present; is usually associated with anisocytosis and an elevation in fetal hemoglobin levels; and is an important diagnostic clue. The bone marrow reveals hypoplasia or aplasia. The diagnosis is confirmed by the demonstration of an increased number of chromosome breaks and rearrangements in peripheral blood lymphocytes. The use of diepoxybutane to stimulate these breaks and rearrangements provides a sensitive assay that is virtually always positive in children with Fanconi anemia, even before the onset of hematologic abnormalities.
Specific Fanconi genes (FANCA, B, C, and others) have been identified and transmission is generally autosomal, although FANCB is on the X chromosome.
 Differential Diagnosis
Differential Diagnosis
Because patients with Fanconi anemia frequently present with thrombocytopenia, the disorder must be differentiated from idiopathic thrombocytopenic purpura (ITP) and other more common causes of thrombocytopenia. In contrast to patients with ITP, those with Fanconi anemia usually exhibit a gradual fall in the platelet count. Counts less than 20,000/μL are often accompanied by neutropenia or anemia. Fanconi anemia may also be manifested initially by pancytopenia, and must be differentiated from acquired aplastic anemia and other disorders, such as acute leukemia. Examination of the bone marrow and chromosome studies of peripheral blood lymphocytes (chromosomal breakage) will usually distinguish between these disorders.
 Complications
Complications
Complications are those related to thrombocytopenia and neutropenia. Endocrine dysfunction may include growth hormone deficiency, hypothyroidism, or impaired glucose metabolism. Persons with Fanconi anemia have a significantly increased risk of developing malignancies, especially acute nonlymphocytic leukemia (800-fold), head and neck cancers, genital cancers, and myelodysplastic syndromes.
 Treatment
Treatment
Attentive supportive care is critical. Patients with neutropenia who develop fever require prompt evaluation and parenteral broad-spectrum antibiotics. Transfusions are important, but should be used judiciously, especially in the management of thrombocytopenia, which frequently becomes refractory to platelet transfusions as a consequence of alloimmunization. Transfusions from family members should be discouraged because of the negative effect on the outcome of bone marrow transplant. At least 50% of patients with Fanconi anemia respond, albeit incompletely, to oxymetholone, and many recommend institution of androgen therapy before transfusions are needed. However, oxymetholone is associated with hepatotoxicity, hepatic adenomas, and masculinization, and is particularly troublesome for female patients.
The definitive treatment is a reduced intensity hematopoietic stem cell transplant, ideally from a human leukocyte antigen (HLA)–identical sibling donor, although matched unrelated and cord transplant may be considered. Before transplant, any prospective sibling donor must be screened for Fanconi anemia.
 Prognosis
Prognosis
Many patients succumb to bleeding, infection, or malignancy in adolescence or early adulthood. Stem cell transplant does not reduce the increased susceptibility for malignancy.
Mehta P, Locatelli F, Stary J, Smith FO: Bone marrow transplantation for inherited bone marrow failure syndromes. Pediatr Clin North Am 2010;57:147–170 [PMID: 20307716].
Younghoon K, D’Andrea AD: Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 2012;122: 3799–3806 [PMID: 23114602].
ACQUIRED APLASTIC ANEMIA
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Weakness and pallor.
Weakness and pallor.
 Petechiae, purpura, and bleeding.
Petechiae, purpura, and bleeding.
 Frequent or severe infections.
Frequent or severe infections.
 Pancytopenia with hypocellular bone marrow.
Pancytopenia with hypocellular bone marrow.
 General Considerations
General Considerations
Acquired aplastic anemia is characterized by peripheral pancytopenia with a hypocellular bone marrow. Approximately 50% of cases in childhood are idiopathic. Other cases are secondary to idiosyncratic reactions to drugs such as phenylbutazone, sulfonamides, nonsteroidal anti-inflammatory drugs (NSAIDs), and anticonvulsants. Toxic causes include exposure to benzene, insecticides, and heavy metals. Infectious causes include viral hepatitis, infectious mononucleosis (Epstein-Barr virus [EBV]), and human immunodeficiency virus (HIV). In immunocompromised children, aplastic anemia has been associated with human parvovirus B19. Immune mechanisms of marrow suppression are suspected in most cases.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Weakness, fatigue, and pallor result from anemia; petechiae, purpura, and bleeding occur due to thrombocytopenia; and fevers due to generalized or localized infections are associated with neutropenia. Hepatosplenomegaly and significant lymphadenopathy are unusual.
B. Laboratory Findings
Anemia is usually normocytic, with a low reticulocyte count. The WBC count is low, with a marked neutropenia. The platelet count is typically below 50,000/μL and is frequently below 20,000/μL. Bone marrow biopsy shows a marked decrease in cellularity typically less than 20% of normal in severe aplastic anemia.
 Differential Diagnosis
Differential Diagnosis
Examination of the bone marrow usually excludes pancytopenia caused by peripheral destruction of blood cells or by infiltrative processes such as acute leukemia, storage diseases, and myelofibrosis. Many of these other conditions are associated with hepatosplenomegaly. Preleukemic conditions also may present with pancytopenia and hypocellular bone marrows. Cytogenetic analysis of the marrow is helpful, because a clonal abnormality may predict the subsequent development of leukemia. Since congenital anomalies may not be apparent in some children with Fanconi anemia, patients with newly diagnosed aplastic anemia should be studied for chromosome breaks and rearrangements in peripheral blood lymphocytes.
 Complications
Complications
Acquired aplastic anemia is characteristically complicated by infection and hemorrhage, which are the leading causes of death. Other complications are those associated with therapy.
 Treatment
Treatment
Comprehensive supportive care is essential. Febrile illnesses require prompt evaluation and usually parenteral antibiotics. Red blood cell (RBC) transfusions alleviate symptoms of anemia. Platelet transfusions may be lifesaving, but they should be used sparingly because many patients eventually develop platelet alloantibodies and become refractory to platelet transfusions.
Immunomodulation, usually with antithymocyte globulin and cyclosporine or tacrolimus, is associated with a high response rate and improved overall survival. However, incomplete response, relapse, and progression to myelodysplasia/leukemia may occur. Hematopoietic stem cell transplant is the treatment of choice for severe aplastic anemia when an HLA-identical sibling donor is available. Because the likelihood of success with transplant is influenced adversely by receipt of transfusions, HLA typing of family members should be undertaken at the time of diagnosis. Increasingly, patients who lack HLA-identical siblings are able to find matched donors through cord blood banks or the National Marrow Donor Program.
 Prognosis
Prognosis
Children receiving early bone marrow transplant from an HLA-identical sibling have a long-term survival rate of greater than 80%. Sustained, complete remissions may be seen in 65%–80% of patients receiving immunosuppressive therapy. However, both therapies are associated with an increased risk of myelodysplastic syndromes, acute leukemia, and other malignancies in long-term survivors.
Korthof ET, Kekassy AN, Hussein AA: Management of acquired aplastic anemia in children. Bone Marrow Transplant 2013;48:191–195 [PMID: 23292240].
Samarasinghe S et al: Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol 2012;157:339–346 [PMID: 22372373].
ANEMIAS
APPROACH TO THE CHILD WITH ANEMIA
Anemia is a relatively common finding, and identifying the cause is important. Even though anemia in childhood has many causes, the correct diagnosis can usually be established with relatively little laboratory cost. Frequently the cause is identified with a careful history. Nutritional causes should be sought by inquiry about dietary intake; growth and development; and symptoms of chronic disease, malabsorption, or blood loss. Hemolytic disease may be associated with a history of jaundice (including neonatal jaundice) or by a family history of anemia, jaundice, gallbladder disease, splenomegaly, or splenectomy. The child’s ethnicity may suggest the possibility of certain hemoglobinopathies or deficiencies of red cell enzymes, such as glucose-6-phosphate dehydrogenase (G6PD). The review of systems may reveal clues to a previously unsuspected systemic disease associated with anemia. The patient’s age is important because some causes of anemia are age related. For example, patients with iron-deficiency anemia (IDA) and β-globin disorders present more commonly at ages 6–36 months than at other times in life.
The physical examination may also reveal clues to the cause of anemia. Poor growth may suggest chronic disease or hypothyroidism. Congenital anomalies may be associated with constitutional aplastic anemia (Fanconi anemia) or with congenital hypoplastic anemia (Diamond-Blackfan anemia). Other disorders may be suggested by the findings of petechiae or purpura (leukemia, aplastic anemia, hemolytic uremic syndrome), jaundice (hemolysis or liver disease), generalized lymphadenopathy (leukemia, juvenile rheumatoid arthritis, HIV infection), splenomegaly (leukemia, sickle hemoglobinopathy syndromes, hereditary spherocytosis, liver disease, hypersplenism), or evidence of chronic or recurrent infections.
The initial laboratory evaluation of the anemic child consists of a complete blood count (CBC) with differential and platelet count, review of the peripheral blood smear, and a reticulocyte count. The algorithm in Figure 30–1 uses limited laboratory information, together with the history and physical examination, to reach a specific diagnosis or to focus additional laboratory investigations on a limited diagnostic category (eg, microcytic anemia, bone marrow failure, pure red cell aplasia, or hemolytic disease). This diagnostic scheme depends principally on the MCV to determine whether the anemia is microcytic, normocytic, or macrocytic, according to the percentile curves of Dallman and Siimes (Figure 30–2).
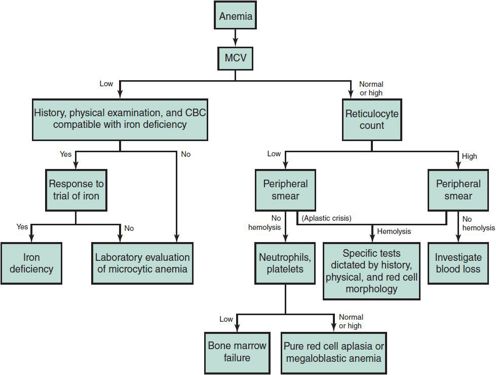
 Figure 30–1. Investigation of anemia.
Figure 30–1. Investigation of anemia.
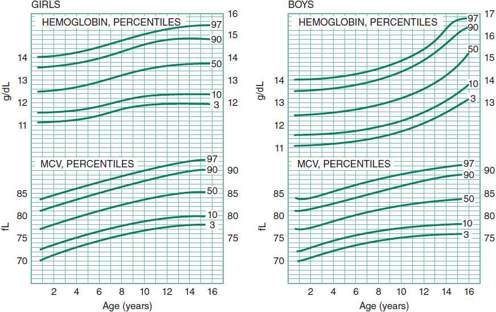
 Figure 30–2. Hemoglobin and red cell volume in infancy and childhood. (Reproduced, with permission, from Dallman PR, Siimes MA: Percentile curves for hemoglobin and red cell volume in infancy and childhood. J Pediatr 1979;94:26.)
Figure 30–2. Hemoglobin and red cell volume in infancy and childhood. (Reproduced, with permission, from Dallman PR, Siimes MA: Percentile curves for hemoglobin and red cell volume in infancy and childhood. J Pediatr 1979;94:26.)
Although the incidence of iron deficiency (ID) in the United States has decreased significantly with improvements in infant nutrition, it remains an important cause of microcytic anemia, especially at ages 6–24 months. A trial of therapeutic iron is appropriate in such children, provided the dietary history is compatible with ID and the physical examination or CBC does not suggest an alternative cause for the anemia. If a trial of therapeutic iron fails to correct the anemia and microcytosis, further evaluation is warranted.
Another key element of Figure 30–1 is the use of both the reticulocyte count and the peripheral blood smear to determine whether a normocytic or macrocytic anemia is due to hemolysis. Typically hemolytic disease is associated with an elevated reticulocyte count, but some children with chronic hemolysis initially present during a period of a virus-induced aplasia when the reticulocyte count is not elevated. Thus, review of the peripheral blood smear for evidence of hemolysis (eg, spherocytes, red cell fragmentation, sickle forms) is important in the evaluation of children with normocytic anemias and low reticulocyte counts. When hemolysis is suggested, the correct diagnosis may be suspected by specific abnormalities of red cell morphology or by clues from the history or physical examination. Autoimmune hemolysis is usually excluded by a negative direct antiglobulin test (DAT). Review of blood counts and the peripheral blood smears of the mother and father may suggest genetic disorders such as hereditary spherocytosis. Children with normocytic or macrocytic anemias, with relatively low reticulocyte counts and no evidence of hemolysis on the blood smear, usually have anemias caused by inadequate erythropoiesis in the bone marrow. The presence of neutropenia or thrombocytopenia in such children suggests the possibility of aplastic anemia, malignancy, or severe folate or vitamin B12 deficiency, and usually dictates examination of the bone marrow.
Pure red cell aplasia may be congenital (Diamond-Blackfan anemia), acquired, and transient (transient erythroblastopenia of childhood); a manifestation of a systemic disease such as renal disease or hypothyroidism; or associated with malnutrition or mild deficiencies of folate or vitamin B12.
Janus J, Moerschel S: Evaluation of anemia in children. Am Fam Physician 2010;15:1462–1471 [PMID: 20540485].
PURE RED CELL APLASIA
Infants and children with normocytic or macrocytic anemia, a low reticulocyte count, and normal or elevated numbers of neutrophils and platelets should be suspected of having pure red cell aplasia. Examination of the peripheral blood smear in such cases is important because signs of hemolytic disease suggest chronic hemolysis complicated by an aplastic crisis due to parvovirus infection. Appreciation of this phenomenon is important because chronic hemolytic disease may not be diagnosed until the anemia is exacerbated by an episode of red cell aplasia and subsequent rapidly falling hemoglobin level. In such cases, cardiovascular compromise and congestive heart failure may develop quickly.
1. Congenital Hypoplastic Anemia (Diamond-Blackfan Anemia)
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Age: birth to 1 year.
Age: birth to 1 year.
 Macrocytic anemia with reticulocytopenia.
Macrocytic anemia with reticulocytopenia.
 Bone marrow with erythroid hypoplasia.
Bone marrow with erythroid hypoplasia.
 Short stature or congenital anomalies in one-third of patients.
Short stature or congenital anomalies in one-third of patients.
 General Considerations
General Considerations
Diamond-Blackfan anemia is a relatively rare cause of anemia that usually presents at 2–3 months of age. To date, mutations of genes encoding ribosomal proteins occurring autosomal dominance has been recognized. Early diagnosis is important because treatment with corticosteroids results in increased erythropoiesis in 80% of patients, thus avoiding the difficulties and complications of long-term chronic transfusion therapy.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Signs and symptoms are generally those of chronic anemia, such as pallor and congestive heart failure. Jaundice, splenomegaly, or other evidence of hemolysis are usually absent. Short stature or other congenital anomalies are present in 50% of patients. A wide variety of anomalies have been described; craniofacial and triphalangeal thumbs are the most common.
B. Laboratory Findings
Diamond-Blackfan anemia is characterized by severe macrocytic anemia and marked reticulocytopenia. The neutrophil count is usually normal or slightly decreased, and the platelet count is normal, elevated, or decreased. The bone marrow usually shows a marked decrease in erythroid precursors but is otherwise normal. In older children, fetal hemoglobin levels are usually increased and there is evidence of persistent fetal erythropoiesis, such as the presence of the i antigen on erythrocytes. In addition, the level of adenosine deaminase in erythrocytes is elevated.
 Differential Diagnosis
Differential Diagnosis
The principal differential diagnosis is transient erythroblastopenia of childhood. Children with Diamond-Blackfan anemia generally present at an earlier age, often have macrocytosis, and have evidence of fetal erythropoiesis and an elevated level of red cell adenosine deaminase. In addition, short stature and congenital anomalies are not associated with transient erythroblastopenia. Lastly, transient erythroblastopenia of childhood usually resolves within 6–8 weeks of diagnosis, whereas Diamond-Blackfan anemia is a lifelong affliction. Other disorders associated with decreased red cell production such as renal failure, hypothyroidism, and the anemia of chronic disease need to be considered.
 Treatment
Treatment
Oral corticosteroids should be initiated at the time of diagnosis. Eight percent of patients respond to prednisone, 2 mg/kg/d, and many who respond subsequently tolerate significant tapering of the dose. Patients who are unresponsive to prednisone require chronic transfusion therapy, which inevitably causes transfusion-induced hemosiderosis and the need for chelation. Bone marrow transplant is an alternative definitive therapy that should be considered for transfusion-dependent patients who have HLA-identical siblings. Unpredictable spontaneous remissions occur in up to 20% of patients.
 Prognosis
Prognosis
The prognosis for patients responsive to corticosteroids is generally good, particularly if remission is maintained with low doses of alternate-day prednisone. Patients dependent on transfusion are at risk for the complications of hemosiderosis. There is an increased risk for the development of solid tumors.
Horos R, vonLindern: Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia. Bri J Haematol 2012;159:514–527 [PMID: 23016900].
2. Transient Erythroblastopenia of Childhood
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Age: 6 months to 4 years.
Age: 6 months to 4 years.
 Normocytic anemia with reticulocytopenia.
Normocytic anemia with reticulocytopenia.
 Absence of hepatosplenomegaly or lymphadenopathy.
Absence of hepatosplenomegaly or lymphadenopathy.
 Erythroid precursors initially absent from bone marrow.
Erythroid precursors initially absent from bone marrow.
 General Considerations
General Considerations
Transient erythroblastopenia of childhood is a relatively common cause of acquired anemia in early childhood. The disorder is suspected when a normocytic anemia is discovered during evaluation of pallor or when a CBC is obtained for another reason. Because the anemia is due to decreased red cell production, and thus develops slowly, the cardiovascular system has time to compensate. Therefore, children with hemoglobin levels as low as 4–5 g/dL may look remarkably well. The disorder is thought to be autoimmune in most cases, because IgG from some patients has been shown to suppress erythropoiesis in vitro.
 Clinical Findings
Clinical Findings
Pallor is the most common sign, and hepatosplenomegaly and lymphadenopathy are absent. The anemia is normocytic, and the peripheral blood smear shows no evidence of hemolysis. The platelet count is normal or elevated, and the neutrophil count is normal or, in some cases, decreased. Early in the course, no reticulocytes are identified. The Coombs test is negative, and there is no evidence of chronic renal disease, hypothyroidism, or other systemic disorder. Bone marrow examination shows severe erythroid hypoplasia initially; subsequently, erythroid hyperplasia develops along with reticulocytosis, and the anemia resolves.
 Differential Diagnosis
Differential Diagnosis
Transient erythroblastopenia of childhood must be differentiated from Diamond-Blackfan anemia, particularly in infants younger than age 1 year. In contrast to Diamond-Blackfan anemia, transient erythroblastopenia is not associated with macrocytosis, short stature, or congenital anomalies, or with evidence of fetal erythropoiesis prior to the phase of recovery. Also in contrast to Diamond-Blackfan anemia, transient erythroblastopenia is associated with normal levels of red cell adenosine deaminase. Transient erythroblastopenia of childhood must also be differentiated from chronic disorders associated with decreased red cell production, such as renal failure, hypothyroidism, and other chronic states of infection or inflammation. As with other single cytopenias, the possibility of malignancy (ie, leukemia) should always be considered, particularly if fever, bone pain, hepatosplenomegaly, or lymphadenopathy is present. In such cases, examination of the bone marrow is generally diagnostic. Confusion may sometimes arise when the anemia of transient erythroblastopenia is first identified during the early phase of recovery when the reticulocyte count is high. In such cases, the disorder may be confused with the anemia of acute blood loss or with hemolytic disease. In contrast to hemolytic disorders, transient erythroblastopenia of childhood is not associated with jaundice or peripheral destruction of red cells.
 Treatment & Prognosis
Treatment & Prognosis
By definition, this is a transient disorder. Some children require red cell transfusions if cardiovascular compromise is present. Resolution of the anemia is heralded by an increase in the reticulocyte count, which generally occurs within 4–8 weeks of diagnosis. Transient erythroblastopenia of childhood is not treated with corticosteroids because of its short course.
NUTRITIONAL ANEMIAS
1. Iron-Deficiency Anemia
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Pallor and fatigue.
Pallor and fatigue.
 Poor dietary intake of iron (ages 6–24 months).
Poor dietary intake of iron (ages 6–24 months).
 Chronic blood loss (age > 2 years).
Chronic blood loss (age > 2 years).
 Microcytic hypochromic anemia.
Microcytic hypochromic anemia.
 General Considerations
General Considerations
Iron deficiency (ID) and iron-deficiency anemia (IDA) are a worldwide concern. ID is defined as a state in which there is insufficient iron to maintain normal physiologic functions such that iron stores (serum ferritin or bone marrow iron content) are reduced. IDA is defined as a hemoglobin more than 2 standard deviations below normal for age and gender, which has developed as a consequence of ID.
Normal-term infants are born with sufficient iron stores to prevent ID for the first 4 months of life, whereas premature infants have reduced iron stores since iron is predominantly acquired in the last trimester. Thus premature infants, as well as those with low birth weight, neonatal anemia, perinatal blood loss, or subsequent hemorrhage may have reduced iron stores. Breast milk is low in iron relative to cow’s milk and fortified formulas, and without iron supplementation, ID may develop in exclusively breast-fed children.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Symptoms and signs vary with the severity of the deficiency. ID is usually asymptomatic. IDA may be associated with, pallor, fatigue, and irritability. A history of pica is common. It is controversial whether or not ID/IDA adversely affects long-term neurodevelopment and behavior. IDA is associated with increased lead absorption and subsequent neurotoxicity.
B. Laboratory Findings
According to the American Academy of Pediatrics (AAP) 2010 guidelines, screening for anemia should be performed at about 12 months of age with determination of hemoglobin concentration and an assessment of risk factors for ID/IDA. Risks include low socioeconomic status, prematurity or low birth weight, lead exposure, exclusive breast-feeding beyond 4 months of age without iron supplementation, weaning to whole milk or complementary foods that do not include iron, feeding problems, poor growth, and inadequate nutrition. If the hemoglobin is less than 11 mg/dL or there is a high risk for ID, an iron evaluation should be performed. There is no single measurement that will document the iron status; recommended tests include serum ferritin and C-reactive protein or reticulocyte hemoglobin concentration.
 Differential Diagnosis
Differential Diagnosis
The differential diagnosis is that of microcytic, hypochromic anemia. The possibility of thalassemia (α-thalassemia, β-thalassemia, and hemoglobin E disorders) should be considered, especially in infants of African, Mediterranean, or Asian ethnic background. In contrast to infants with ID, those with thalassemia generally have an elevated red cell number and are less likely, in mild cases, to have an elevated RBC distribution width (the index of the MCV divided by the red cell number is usually < 13). Thalassemias are associated with normal or increased levels of serum iron and ferritin and with normal iron-binding capacity. The hemoglobin electrophoresis in β-thalassemia minor typically shows an elevation of hemoglobin A2 levels, but coexistent ID may lower the percentage of hemoglobin A2 into the normal range. Hemoglobin electrophoresis will also identify children with hemoglobin E, a cause of microcytosis common in Southeast Asians. In contrast, the hemoglobin electrophoresis in α-thalassemia trait is normal. Lead poisoning has also been associated with microcytic anemia, but anemia with lead levels less than 40 mg/dL is often due to coexistent ID.
The anemia of chronic inflammation or infection is normocytic but in late stages may be microcytic. This anemia is usually suspected because of the presence of a chronic systemic disorder and an elevated CRP. Relatively mild infections, particularly during infancy, may cause transient anemia. Thus, screening tests for anemia should not be obtained within 3–4 weeks of such infections.
 Treatment
Treatment
The AAP has published guidelines for routine iron intake for children. If a child has a hemoglobin of 10–11 mg/dL at the 12-month screening visit, the child can be closely monitored or empirically treated with iron supplementation with a recheck of hemoglobin in one month.
If a child is found to have ID/IDA, the recommended oral dose of elemental iron is 6 mg/kg/d in three divided daily doses. Parenteral administration of iron is rarely necessary. Iron therapy results in an increased reticulocyte count within 3–5 days, which is maximal between 5 and 7 days. The rate of hemoglobin rise is inversely related to the hemoglobin level at diagnosis. Resolution of the anemia is within 4–6 weeks. Treatment is generally continued for a few additional months to replenish iron stores.
Baker RD, Greer FR; the Committee on Nutrition: Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics 2010;126:1040–1050 [PMID: 2093825].
Eden AN, Sandoval C: Iron deficiency in infants and toddlers in the United States. Pediatr Hematol Oncol 2012;29:704–709 [PMID: 2303474].
2. Megaloblastic Anemias
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Pallor and fatigue.
Pallor and fatigue.
 Nutritional deficiency or intestinal malabsorption.
Nutritional deficiency or intestinal malabsorption.
 Macrocytic anemia.
Macrocytic anemia.
 Megaloblastic bone marrow changes.
Megaloblastic bone marrow changes.
 General Considerations
General Considerations
Megaloblastic anemia is a macrocytic anemia caused by deficiency of cobalamin (vitamin B12), folic acid, or both. Cobalamin deficiency due to dietary insufficiency may occur in infants who are breast fed by mothers who are strict vegetarians or who have pernicious anemia. Intestinal malabsorption is the usual cause of cobalamin deficiency in children and occurs with Crohn disease, chronic pancreatitis, bacterial overgrowth of the small bowel, infection with the fish tapeworm (Diphyllobothrium latum), or after surgical resection of the terminal ileum. Deficiencies due to inborn errors of metabolism (transcobalamin II deficiency and methylmalonic aciduria) also have been described. Malabsorption of cobalamin due to deficiency of intrinsic factor (pernicious anemia) is rare in childhood.
Folic acid deficiency may be caused by inadequate dietary intake, malabsorption, increased folate requirements, or some combination of the three. Folate deficiency due to dietary deficiency alone is rare but occurs in severely malnourished infants and has been reported in infants fed with goat’s milk not fortified with folic acid. Folic acid is absorbed in the jejunum, and deficiencies are encountered in malabsorptive syndromes such as celiac disease. Anticonvulsion medications (eg, phenytoin and phenobarbital) and cytotoxic drugs (eg, methotrexate) also have been associated with folate deficiency, caused by interference with folate absorption or metabolism. Finally, folic acid deficiency is more likely to develop in infants and children with increased requirements. This occurs during infancy because of rapid growth and also in children with chronic hemolytic anemia. Premature infants are particularly susceptible to the development of the deficiency because of low body stores of folate.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Infants with megaloblastic anemia may show pallor and mild jaundice as a result of ineffective erythropoiesis. Classically, the tongue is smooth and beefy red. Infants with cobalamin deficiency may be irritable and may be poor feeders. Older children with cobalamin deficiency may complain of paresthesias, weakness, or an unsteady gait and may show decreased vibratory sensation and proprioception on neurologic examination.
B. Laboratory Findings
The laboratory findings of megaloblastic anemia include an elevated MCV and mean corpuscular hemoglobin (MCH). The peripheral blood smear shows numerous macro-ovalocytes with anisocytosis and poikilocytosis. Neutrophils are large and have hypersegmented nuclei. The white cell and platelet counts are normal with mild deficiencies but may be decreased in more severe cases. Examination of the bone marrow is not indicated, but it typically shows erythroid hyperplasia with large erythroid and myeloid precursors. Nuclear maturation is delayed compared with cytoplasmic maturation, and erythropoiesis is ineffective. The serum indirect bilirubin concentration may be slightly elevated.
Children with cobalamin deficiency have a low serum vitamin B12 level, but decreased levels of serum vitamin B12 may also be found in about 30% of patients with folic acid deficiency. Negative results should not negate treatment if clinically compatible symptoms are present. The level of red cell folate is a better reflection of folate stores than is the serum folic acid level. Elevated serum levels of metabolic intermediates (methylmalonic acid and homocysteine) may help establish the correct diagnosis. Elevated methylmalonic acid levels are consistent with cobalamin deficiency and generally decrease with treatment, whereas elevated levels of homocysteine occur with both cobalamin and folate deficiency.
 Differential Diagnosis
Differential Diagnosis
Most macrocytic anemias in pediatrics are not megaloblastic. Other causes of an increased MCV include drug therapy (eg, anticonvulsants, anti-HIV nucleoside analogues), Down syndrome, an elevated reticulocyte count (hemolytic anemias), bone marrow failure syndromes (Fanconi anemia, Diamond-Blackfan anemia), liver disease, and hypothyroidism.
 Treatment
Treatment
Treatment of cobalamin deficiency due to inadequate dietary intake is readily accomplished with high-dose oral supplementation that is as effective as parenteral treatment if absorption is normal. Folic acid deficiency is treated effectively with oral folic acid in most cases. Children at risk for the development of folic acid deficiencies, such as premature infants and those with chronic hemolysis, are often given folic acid prophylactically.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


