Fig. 3.1
Perifollicular hypopigmentation is due to accumulated keratin at follicular orifices
Melanin is the principal pigment present in the skin that looks darker the more concentrated it is. It can be found in two different colors: yellow/red (pheomelanin) and brown/black (eumelanin). Differences in skin pigmentation among individuals are related to the concentration and ratio between eumelanin and pheomelanin, as well as the distribution of melanin in the basal layer and epidermis, rather than the number of melanocytes as its number is relatively constant in different races. In most East Asians, Indians, and Africans, the predominant pigment is eumelanin, and among the fair red-headed Caucasians, the predominant pigment is pheomelanin.
Besides the melanin concentration and ratio, its location at the epidermis or dermis also affects the final skin color due to Tyndall effect. Longer wavelengths such as red penetrate deeper and are absorbed by melanin. Since blue does not penetrate so deeply it is not absorbed and is reflected back, which is why dermal pigment appears blue in dermal melanocytosis.
Melanocytes are the key player in most pigmentary disorders. They are dendritic cells that are derived from melanoblasts, which originate from the neural crest. During embryogenesis, progenitor melanoblasts migrate between mesodermal and ectodermal layers to reach their final destinations in the epidermis and hair follicular bulbs, as well as the inner ear cochlea, choroids, ciliary body, and iris [2]. Any disruption of the migration of melanoblasts from the neural crest to the basal layer of the epidermis, lack or excess in production of melanin, and transfer of melanosomes from melanocytes to keratinocytes in the epidermis will result in pigmentary defects with variable degrees of extracutaneous involvement. Therefore, the eyes and hearing in addition to the skin and hair are defective in some genetic pigmentary disorders, e.g., Waardenburg syndrome. Melanin biosynthesis is primarily regulated by tyrosinase, a copper-dependent enzyme that converts tyrosine to dihydroxyphenylalanine (DOPA). It is the most important rate-limiting enzyme in melanogenesis.
Normal Variants in Skin Pigmentation
Knowledge about normal variations in skin color is important for us to distinguish them from other abnormal skin discolorations to avoid overdiagnosis. Below are some of the common variants among East Asian children.
1.
Pigmentary demarcation lines (PDL)
Pigmentary demarcation lines (PDL), also known as Futcher’s lines or Voight’s lines, were first described by the Viennese anatomist Christian A. Voight.
In all races, the dorsal skin surfaces have relatively higher pigmentation compared to the ventral surfaces. There are lines of demarcation (Type A–E) between dorsal and ventral skin surfaces (Figs. 3.2, 3.3, and 3.4) [3]. These demarcation lines are bilateral symmetrical/midline and present from infancy and persist throughout adulthood.
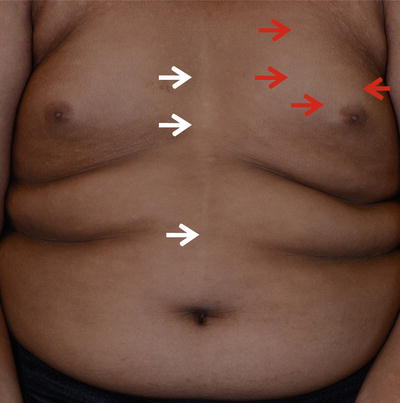
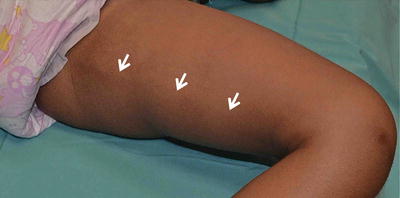
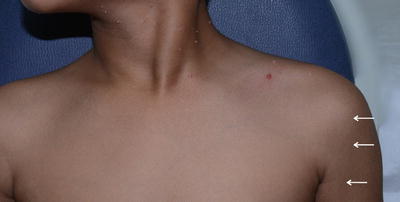
Fig. 3.2
Type C and type E pigment demarcation lines. Type C—hypopigmented lines present as paired median or paramedian lines on the chest with midline abdominal extension (white arrow ). Type E—Bilateral chest markings (hypopigmented macules and patches) in a zone that runs from the mid third of the clavicle to the periareolar skin (red arrow)
Fig. 3.3
Type B pigment demarcation lines. A curved line on the posteromedial thigh that extends from perineum to popliteal fossa (white arrow )
Fig. 3.4
Type A pigment demarcation lines. A vertical line along the anterolateral portion of upper arm that may extend into the pectoral region (white arrow )
Knowledge about them is essential; i.e., Type C and E pigmentary demarcation lines may be confused with nevus depigmentosus.
3.
Linea nigra (Fig. 3.5)
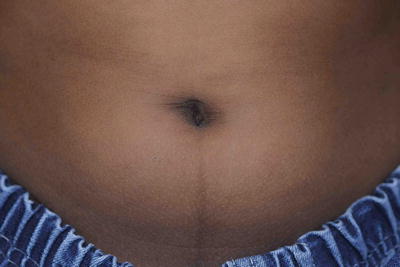
Fig. 3.5
Linea nigra. It is a fairly common finding in children especially those with darker skin types
Other darkly pigmented areas that are regarded as normal include the genital area, the elbows and the knees, the knuckles, and in many, a mild degree of infraorbital pigmentation.
Classification of Pigmentary Disorders in Children
Although most pigmentation disorders are benign or nonspecific, some pigmentary disorders present cosmetic or psychological challenges to the patient, necessitating systematic evaluation and treatment. Others may be indicators of underlying genetic disorders with systemic involvement.
Pigmentary disorders are first broadly classified into three groups (Fig. 3.7):
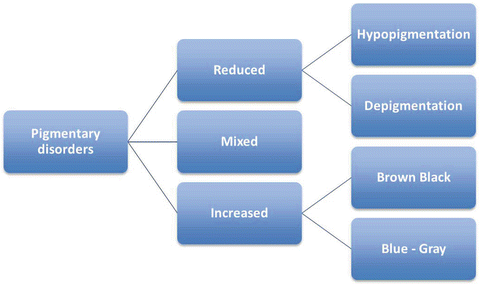
Fig. 3.7
Classification of pigmentary disorders in children
1.
2.

Hypopigmentation disorders in children can be due to a wide variety of congenital and acquired diseases (Table 3.1).
Table 3.1
Causes of hypopigmentation in children
Hypopigmentary disorders in children | |
|---|---|
Group A (Localized with early onset) | Group A-1: Hypopigmented 1. Nevus depigmentosusa 2. Hypomelanosis of Itoa 3. Nevus anemicus 4. Tuberous sclerosis 5. Post inflammatory hypopigmentationa Group A-2: Depigmented 1. Piebaldism 2. Waardenburg syndrome 3. Vitiligoa |
Group B (Generalized and early onset) | Group B-1: Skin, hair and eyes 1. Oculocutaneous albinism 2. Chediak–Higashi syndrome 3. Hermansky–Pudlak syndrome 4. Prader Willi and Angelman syndrome 5. Metabolic disorders (a) Phenylketonuria (b) Histidinemia (c) Homocystinuria Group B-2: Skin and hair only 1. Griscelli syndrome 2. Elejalde syndrome 3. Menkes disease 4. Copper and Selenium deficiency |
Group C/D (Late onset localized/generalized) | Depigmentated 1. Vitiligob 2. Post inflammatory leukoderma Hypopigmentation 1. Post inflammatory hypopigmentation (a) Eczema or psoriasis (b) Pityriasis lichenoides chronica (c) Lichen striatus (d) Infection 2. Pityriasis alba 3. Hypopigmented mycosis fungoides 4. Morphea 5. Lichen sclerosus et atrophicus (LSEA) |
Since their underlying causes are heterogeneous and histological examination of the skin alone is rarely diagnostic, a systematic clinical approach is essential (Fig. 3.14).
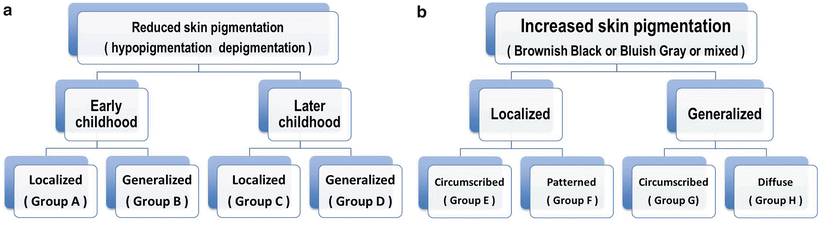
Fig. 3.14
(a, b) Classification of hypopigmentation and hyperpigmentation in children
The disorders are usually classified based on the age of onset into early and later childhood, and in each category they are subdivided into localized and generalized hypopigmentation. Other clinical findings, listed in Table 3.2, are helpful to distinguish the disorders further.
Table 3.2
Helpful clinical findings to subcategorize children with hypopigmentation disorder
Helpful clinical findings | |
|---|---|
Age of onset | (A) Early onset: At birth to first 2 years of life (B) Late onset: After 2 year-old |
Distribution and pattern | (A) Involvement of skin ± hair ± mucous membranes ± eyes (B) Localized Generalized (a) Diffuse (b) Circumscribed (C) Patterned or non-patterned |
Morphology | Surface changes 1. Scaly 2. Atrophy 3. Verrucous |
Extracutaneous features | Neurological, musculoskeletal, eyes and ears, etc. |
Others | Special tests 1. Wood’s light 2. Test for skin sensation Family tree to identify the pattern of inheritance |
Similarly, hyperpigmentation disorders in children can be due to many causes (Table 3.3). Localized or patterned hyperpigmentation of early onset is frequently developmental or hereditary in origin. However, pigmented lesions may also be acquired later in childhood following inflammatory dermatoses especially among Asian children with skin types IV to VI.
Table 3.3
Causes of hyperpigmentation and dyschromia in children
Hyperpigmentary disorders in children | |
|---|---|
Group E: Localized circumscribed hyperpigmentation (single to few lesions) | Group E-1: Brown/black 1. Café au lait maculea 2. Congenital melanocytic nevusa 3. Segmental pigmentation disorder 4. Nevus spilus 5. Becker’s nevus 6. Post inflammatory hyperpigmentation Group E-2: Blue gray 1. Mongolian spots 2. Nevus of Ota 3. Nevus of Ito 4. Phakomatosis pigmentovascularisa 5. Ashy dermatosesa |
Group F: Localized patterned hyperpigmentation | 1. Epidermal nevus 2. Hyperpigmentation stage of IP 3. Linear and whorled nevoid hypermelanosis 4. X linked chondrodysplasia punctata |
Group G: Generalized circumscribed hyperpigmentation (few to multiple) | Group G-1: Brown/black 1. Maculopapular cutaneous mastocytosis 2. Multiple lentigines syndrome 3. Post inflammatory hyperpigmentation 4. Transient neonatal pustular melanosis 5. Giant congenital melanocytic nevi with satellite lesions 6. Tinea versicolor 7. Xeroderma pigmentosum (early) Group G-2: Blue-gray 1. Ashy dermatoses 2. Phakomatosis pigmentovascularis 3. Generalized fixed drug eruption |
Group H: Generalized diffuse hyperpigmentation | Group H-1: Without skin thickening or hyperkeratosis 1. Drugs e.g., clofazimine, minocycline 2. Post inflammatory hyperpigmentation 3. Chronic liver failure and renal failure 4. Hemochromatosis Group H-2: With skin thickening or hyperkeratosis 1. Congenital ichthyosis 2. Acanthosis nigrican 3. Generalized exfoliative dermatitis |
Group I: Dyschromatosis (mixed hypo- and hyperpigmentation | 1. Dyschromatosis symmetrica hereditaria 2. Dyschromatosis universalis hereditaria 3. Early xeroderma pigmentosum 4. Chronic arsenic poisoning 5. Dyschromic amyloidosis cutis |
Epidermal melanosis is usually brown to black in color and is enhanced with a Wood’s lamp, whereas dermal melanosis tends to produce blue-gray lesions and is not enhanced by Wood’s light. Some disorders, such as melasma in adults, may have dermal and epidermal changes and can be classified as mixed.
The dyschromatoses are a group of pigmentary disorders characterized by a combination of hyper- and hypopigmentation without atrophy or telangiectasia as seen in poikiloderma.
Group A-1: Early-Onset Hypopigmentary Disorder
Nevus Depigmentosus
Introduction
Nevus depigmentosus (ND) is defined as a congenital nonprogressive hypopigmented macule or patch that was first reported by Lesser in 1884. It is caused by aberrant transfer of melanosomes to the keratinocytes.
Clinical Features
Morphology
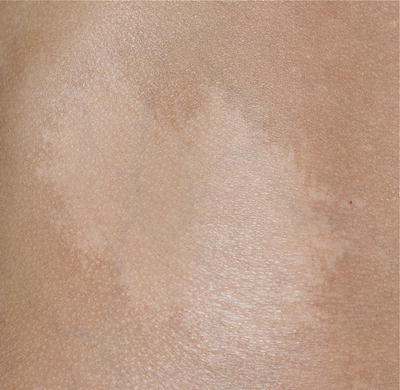
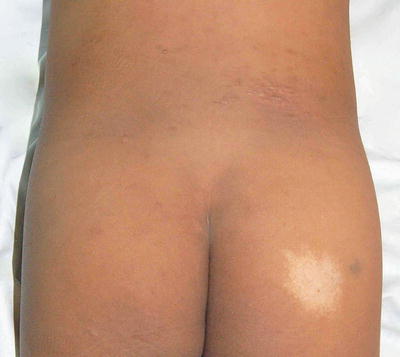
Nevus depigmentosus is a misnomer as the area of involvement is actually hypopigmented and not depigmented patch. It has an irregular but well-defined margin. The shape of the lesion is variable and may be round or rectangular. Its surface is smooth and non-scaly (Fig. 3.15).
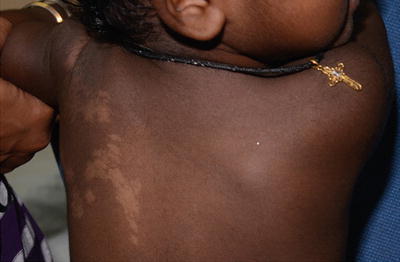
Fig. 3.15
Nevus depigmentosus. A healthy baby girl was noted to have a hypopigmented patch on the left upper back since birth
Distribution and Pattern
It may occur at most body sites, but the most common site is the trunk [4]. Occasionally, a patient will have a unilateral segmental lesion that follows the lines of Blaschko. Due to clinical overlap among some of these lesions with hypomelanosis of Ito, the umbrella term of “nevoid linear hypopigmentation” is used by some clinicians.
Clinical diagnostic criteria for nevus depigmentosus proposed by Coupe in 1976 are:
1.
Leukoderma present at birth or onset early in life
2.
No alteration in distribution of leukoderma throughout life
3.
No alteration in texture, or change of sensation, in the affected area
4.
No hyperpigmented border around the achromic area
Extracutaneous Findings
It has rarely been reported in association with hemihypertrophy and neurological deficit.
Clinical Course
Nevus depigmentosus is usually present at birth or becomes evident shortly thereafter. Its size increases in proportion to the growth of the child. In Korea, a clinical survey that involved 67 patients with nevus depigmentosus has concluded that the majority (92.5 %) of them present before 3 years of age, but some lesions also appeared later in childhood (7.5 %). Forty patients (59.7 %) had the isolated type of nevus depigmentosus and 27 patients (40.3 %) had the segmental type [5].
Differential Diagnosis
1.
Vitiligo
Cases of late-onset nevus depigmentosus are sometimes misdiagnosed as segmental vitiligo. It is important to distinguish it from childhood vitiligo because they have different prognostic and psychosocial effects. Furthermore, ND is not responsive to topical steroid and phototherapy.
2.
Nevus anemicus (NA)
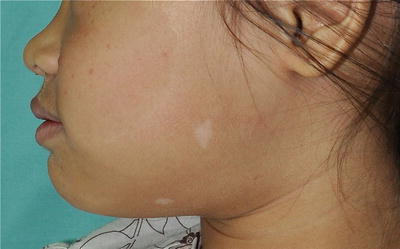
Nevus anemicus typically presents with an asymptomatic pale macule or patch with irregular margin that has been present since birth and grows with the child. It may be seen in close association with a port-wine stain (Fig. 3.16). It is due to persistent increase in vascular tone, which results in localized vasoconstriction.
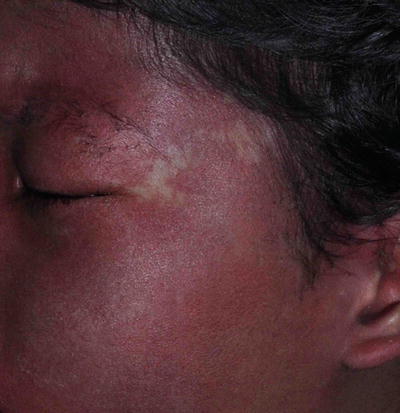
Fig. 3.16
Nevus anemicus. The boy has a congenital pale patch on his left temple with underlying portwine stain
Three simple maneuvers to differentiate NA from others are:
(a)
Under diascopy, the lesion becomes indistinguishable from the blanched surrounding normal tissue
(b)
Wood lamp doesn’t accentuate the lesion
(c)
Rubbing causes erythema of the surrounding area but not within the lesion itself.
3.
Hypomelanosis of Ito
4.
Piebaldism
5.
Tuberous sclerosis
6.
Post-inflammatory hypopigmentation (Fig. 3.17)
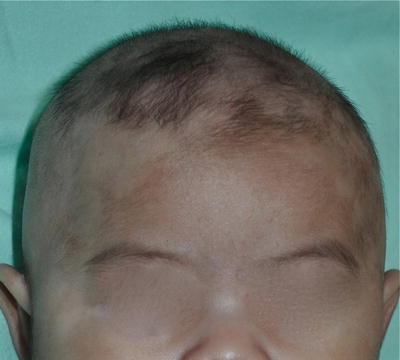
Fig. 3.17
Post-inflammatory hypopigmentation on scalp and hairlines. Noted in a 3-month-old boy secondary to infantile seborrheic dermatitis
Treatment: Camouflage is helpful if the lesion occurs on cosmetically important areas. Sun protection with clothing and sunscreen is useful to prevent sunburn. Autologous melanocytic transplantation is another option that has given variable results [6].
Hypomelanosis of Ito
Introduction
Hypomelanosis of Ito (HI) is a descriptive term applied to individuals with skin hypopigmentation along the lines of Blaschko. Even though originally described as a purely cutaneous disease by Dr. Ito in 1952, subsequent reports have showed a frequent association with neurological abnormalities leading to frequent characterization as a neurocutaneous disorder. Hypomelanosis of Ito is believed to be due to chromosomal mosaicism and sporadic mutations. It is not an inherited disorder as the chromosomal defect occurs after conception. Recurrence is uncommon. The specific gene(s) involved has not been confirmed.
Clinical Features
Morphology
Lesions first appear at birth or become apparent within the first 2 years of life as small hypopigmented macules that are arranged in linear, whorls, or streaks. Their surfaces are smooth, non-scaly, and not preceded by inflammation. Wood’s lamp examination is helpful for Asians with fair skin and also during the early infantile period as skin pigmentation according to skin type has not been established.
Distribution and Pattern
The hypopigmented macules are arranged along the Blaschko lines (Figs. 3.18 and 3.19)
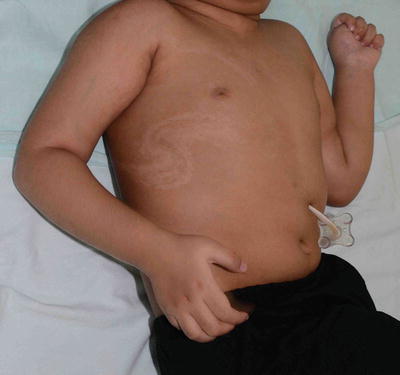
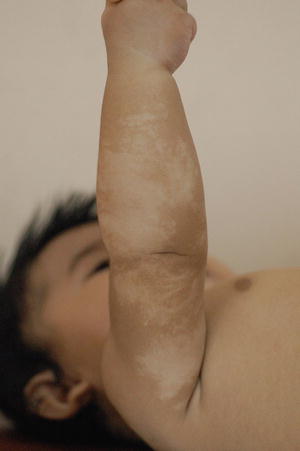
Fig. 3.18
Hypomelanosis of Ito. Hypopigmentation is distributed in swirls along the lines of Blaschko in this girl with neurological defect. The pigmentary disturbance is often noticed soon after birth and is non-progressive
Fig. 3.19
Hypomelanosis of Ito. Hypopigmentation is distributed in linear fashion along the lines of Blaschko in this infant since birth
It may be unilateral or bilateral. Palmoplantar regions, scalp, and mucous membranes are usually not affected.
Extracutaneous Features
Extracutaneous involvement is variable, and includes central nervous system, musculoskeletal, dental, eye, and cardiac abnormalities.
The literature contains reviews, mostly from neurology departments that report rates of hypomelanosis of Ito-associated neurologic abnormalities as high as 75–94 %.
This figure is higher in a pediatric neurology clinic-generated series and when systemic involvement was used as a key diagnostic criterion [7, 8]. More recent groups estimate that the associated anomaly rate is 30–50 %.
Affected children require a careful medical, neurologic, ocular, and musculoskeletal examination and a regular developmental milestones assessment.
Further imaging studies are tailored to the clinical findings.
Common findings include seizure, mental retardation, hearing and visual abnormalities, and tooth and musculoskeletal defects.
Diagnosis of Hypomelanosis of Ito
Some authorities advocate stricter criteria for diagnosis, requiring cutaneous involvement of two or more segments plus systemic involvement for definite diagnosis of hypomelanosis of Ito [9]. However, these criteria cannot be considered to be definitive until the etiology is more clearly understood as there are a lot of clinical overlaps, e.g., systematized nevus depigmentosus. Hence, the distinction between hypomelanosis of Ito and nevus depigmentosus may be artificial.
Hypomelanosis of Ito is a clinical description and not a diagnosis. Practically, nevoid hypopigmentation with or without systemic involvement is probably a better way to approach these skin lesions.
Differential diagnoses are the hypopigmented stage of incontinentia pigmenti, linear whorled nevoid hypermelanosis, Goltz syndrome, and systematized nevus depigmentosus.
Cytogenetic analysis of peripheral blood lymphocytes and skin fibroblasts should be considered in all the children with segmental or linear pigmentary disorders with extracutaneous involvement to look for chromosomal mosaicism.
Treatment
No specific treatment is available. Camouflage is helpful for areas of cosmetic concern.
And sunblock is useful to reduce the pigmentary differences as the lesions will be more obvious with the differential tanning response compared with the surrounding normal skin.
Tuberous Sclerosis Complex
Introduction
The name “tuberous sclerosis” comes from “tubers” (protuberances) and areas of “sclerosis” (hardening) in the cerebral gyri that calcify with age. Tuberous sclerosis complex (TSC) was first recognized by Friedrich Daniel von Recklinghausen in 1862.
TSC is characterized by the formation of hamartomas in various organs, e.g., the brain, heart, lung, kidneys, and skin. It is an autosomal dominant disorder with almost complete penetrance but a wide range of clinical severity even among the siblings. Spontaneous mutation accounts for 66–86 % of cases [10, 11]. It has no predilection for gender.
The estimated prevalence of TSC is around one case for every 6,000–10,000 births [12]. A study in Taiwan revealed that the prevalence of TSC was estimated to be 1:95,136 and the prevalence for cases less than 6 years of age was 1:14,608 [13]. However, its true incidence is not known because of a number of undiagnosed cases consisting mostly of mildly affected or asymptomatic individuals.
TSC is caused by mutations of either the TSC1 gene on chromosome 9q34 encoding hamartin or the TSC2 gene on chromosome 16p13 encoding tuberin. Patients with TSC1 mutations generally have milder disease than patients with TSC2 mutation. The tuberous sclerosis gene products, hamartin and tuberin, form a tumor suppressor complex which drives Rheb (Ras homologue enriched in brain) into the inactive guanosine diphosphate-bound state.
Clinical Features
Among the diagnostic criteria for tuberous sclerosis complex, four major and one minor criteria are manifested on the skin.
1.
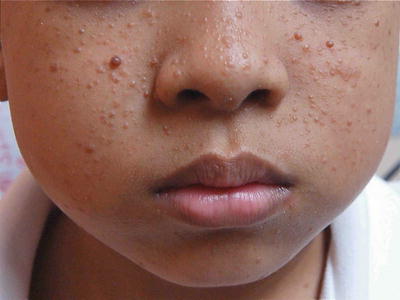
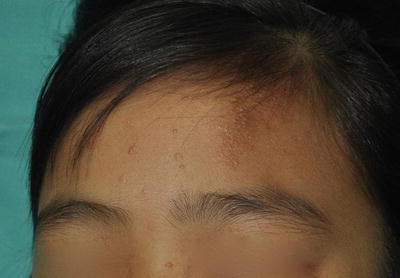
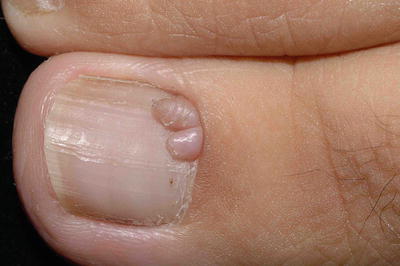

4.
5.
Hypomelanotic lesions in “Confetti” pattern (minor)
The skin findings have important diagnostic value because they are common and readily identifiable by routine physical examination. They may be the only clues that the dermatologist has to the diagnosis of TSC. The prevalence and age of onset of these findings are listed in Table 3.4 [14].
Table 3.4
Skin findings of tuberous sclerosis complex
Age of onset | Prevalence | |
|---|---|---|
Hypomelanotic macules | Usually at birth or infancy | 97.2 % |
Facial angiofibroma | 2–5 year-old | 74.5 % |
Shagreen patch | Rarely at birth. Increase its size with age | 48.1 % |
Forehead fibrous plaque | Can be seen at birth or early infancy | 18.9 % |
Periungual fibroma | Present at puberty or soon after | 15.1 % |
Hypopigmented Skin Lesions in TSC
Hypopigmented macules or patches are the earliest sign of TSC and are present in up to 90 % of patients.
Morphology: The macules and patches in TSC are hypopigmented, well defined with non-scaly surface. Its shapes vary from polygonal to ash leaf or arranged in confetti pattern.
Distribution and pattern: These lesions are found mainly over the body and extremities.
Clinical course: Hypopigmented skin lesions in TSC are usually detected at birth and their size increases according to body size.
Differential Diagnosis
A single hypopigmented macule is common and has been described in 0.2–0.3 % of all neonates [15]. They need to be differentiated from those listed in group A(1).
Other major findings as stated below are not readily available by physical examination and often need radiological imaging for detection and confirmation.
Cortical tuber, subependymal nodule, subependymal giant cell astrocytoma
Cardiac rhabdomyoma, single or multiple
Lymphangiomyomatosis
Renal angiomyolipoma
The classic diagnostic triad of seizures, mental retardation, and facial angiofibromas is evident in only 29 % of cases; 6 % of TSC patients have none of these three findings [16].
Management
Management is multidisciplinary and individualized based on their severity and organs involved. As TSC has variable penetrance and its clinical signs are progressive, regular follow-up is mandatory as certain signs appear later in life. Camouflage and photoprotection is beneficial for the hypopigmented lesions. Carbon dioxide, pulsed dye laser, and topical sirolimus/rapamycin [17] are helpful for facial angiofibromas, but slowly recur once the treatment stopped.
Genetic counseling is helpful, but its benefit is often limited by its wide variation in genetic expression and the high frequency of spontaneous gene mutation in TSC. A careful family history, skin examination, and relevant imaging of other family members are recommended.
Group A-2: Early-Onset Depigmentary Disorder
Piebaldism
Introduction
Piebaldism is a rare autosomal dominant disorder characterized by congenital poliosis and leukoderma. It results from mutations of the c-kit gene on chromosome 4q11–12 [18].
A mutation in the c-kit proto-oncogene results in abnormal tyrosine kinase transmembrane cellular receptors and causes abnormal melanocyte embryogenesis with defective melanoblast proliferation, migration, and distribution. There is absence of melanocytes and melanin production within the depigmented lesion.
Clinical Features
Morphology
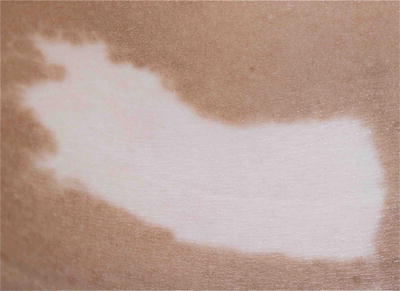
The areas of depigmented white patches are irregular, well-demarcated, and milky white in color. There is presence of islands of normal pigmented and hyperpigmented macules within these depigmented patches. This is typical and helpful in the clinical diagnosis.
Distribution and Pattern
Skin
The depigmented skin patches have a characteristic distribution pattern that favor the forehead, central chest and abdomen, upper arms, and lower legs. Posterior midline, hands, and feet are usually spared.
Hair
A white forelock which consists of a tuft of white hair over the midfrontal scalp is present in 80–90 % of patients with depigmentation of the underlying scalp (Fig. 3.26) [19]. However, its absence does not exclude the diagnosis. Poliosis of eyebrows and eyelashes may be seen.
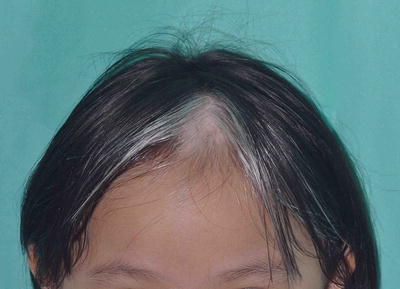
Fig. 3.26
Piebaldism. The white forelock is characteristic of piebaldism. White patches in piebaldism are present from birth and is a useful distinguishing feature from vitiligo. However, her eyebrows and eyelashes are not involved in this girl
Clinical Course
The lesions are present at birth but remain constant throughout life, although some variability in pigmentation may occur with sun exposure.
Differential Diagnosis (Table 3.5)
Table 3.5
Clinical features of piebaldism, early onset vitiligo and Waardenburg’s syndrome
Piebaldism | Vitiligo | Waardenburg’s syndrome | |
|---|---|---|---|
Morphology | Depigmentation Well defined With areas of normal or hyperpigmentation | Depigmentation Well defined | Depigmentation |
Distribution | |||
Skin | Central forehead mid arms/legs Sparing hands/feet | Periorificial | Face, neck, body and dorsal limbs |
Hair/eyebrow | Poliosis | Poliosis | Poliosis |
Mucosal | Spared | Present | Present |
Clinical course | Present at birth Non-progressive | At any age, but rarely at birth Usually progressive | Present at birth Non-progressive |
Extracutaneous | Usually isolated | Autoimmune conditions e.g., thyroiditis | Heterochromia irides Deafness |
Histology | Absence of melanocyte | Absence of melanocytes | Absence of melanocytes |
1.
Vitiligo
Morphologically, vitiligo and piebaldism are similar with well-demarcated depigmented patches. But the presence of stable depigmented patches since birth, family history, the characteristic distribution pattern, and islands of normal or hyperpigmented macules within the areas of depigmented patches allow the differentiation of piebaldism from vitiligo.
2.
Waardenburg syndrome (WS)
WS needs to be considered for all children with piebaldism. Ocular and hearing assessment is mandatory. Piebaldism (Fig. 3.27), facial dysmorphism, heterochromia of the irides, and sensorineural deafness are the main features of Waardenburg syndrome. WS has been divided into four variants (WS1–WS4). Both WS1 and WS2 are transmitted as autosomal dominant conditions with interfamilial and intrafamilial variability. Two far rarer variants WS 3 and 4 include features of WS in association with severe contractures and Hirschsprung disease, respectively [22, 23].
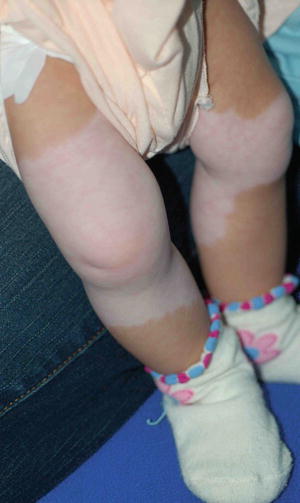
Fig. 3.27
Waardenburg syndrome. The depigmented skin patches have a characteristic distribution pattern that involved mid portion of both lower legs since birth. Both feet are spared. He had a white forelock, lateral displacement of inner canthi (inner canthal distance divided by the interpupillary distance is >0.6) and hearing impairment
Treatment of Piebaldism
Most treatments (e.g., topical calcineurin inhibitor, topical corticosteroid, and phototherapy) are not efficacious. Photoprotection of the depigmented areas is important to protect against sunburn and skin cancer. Cosmetic camouflage can be recommended and helpful. Autologous cultured melanocyte grafts may be worthwhile in selected patients, but this often requires multiple sessions.
Group B: Early-Onset Generalized Diffuse Hypopigmentation
Generalized diffuse hypopigmentation of early onset describes those extensive hypopigmentary disorders that occur from birth up to the first 2 years of life. As neonates often have a lighter skin at birth, some of these congenital hypopigmented lesions can easily go unnoticed until later in infancy. In addition to age of onset, they can be further subcategorized into with or without ocular involvement (Table 3.1).
Diffuse and early-onset hypopigmentation in children often has an underlying genetic cause. Hence, a detailed family tree is mandatory. In all of the disorders outlined in group B, the epidermis generally contains normal numbers of melanocytes and histological findings under light microscope are often not helpful in differentiating these conditions. The pathophysiological defect of hypopigmentation lies in either melanin biosynthesis or melanosome formation and trafficking.
Group B-1: Early-Onset Generalized Diffuse Hypopigmentation with Skin, Hair, and Eyes Involvement
Oculocutaneous Albinism
General features of albinism are as below
(A)
It is a heterogeneous group of pigmentary disorder manifested by generalized hypopigmentation or depigmentation of the skin, eyes, and hair that has onset at birth (Figs. 3.28, 3.29, and 3.30)
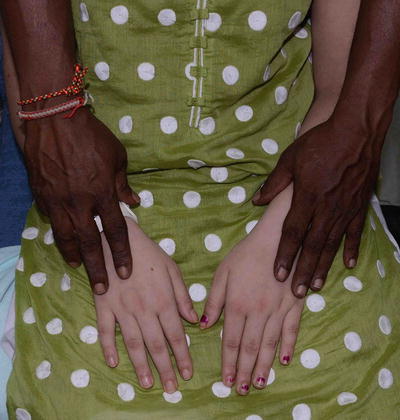
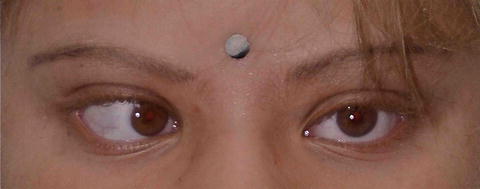
Fig. 3.28
Albinism. Hands of the patient and her father
Fig. 3.29
Albinism
Fig. 3.30
Albinism and hypochromic iris
In oculocutaneous albinism (OCA) type 1A, there is permanent and complete absence of pigment from birth. In OCA types 1B, 2, 3, and 4, however, pigment production may increase over time.
(B)
Historically, OCA is divided only into two clinical types: tyrosinase positive or negative. Advances in molecular genetics and better understanding of genotype–phenotype correlation have given rise to a more accurate classification of Albinism into seven groups.
(C)
All of them are autosomal recessive. Both sexes are equally involved.
(D)
In OCA, epidermal melanocytes are present in normal amount and distribution but do not synthesize adequate melanin due to tyrosinase defects or melanosomal dysfunction.
(E)
Overall, an estimated 1 in 20,000 people worldwide are born with oculocutaneous albinism. The condition affects people in many ethnic groups and geographical regions. Types 1 and 2 are the most common forms of this condition; types 3 and 4 are less common. However, particular types of albinism are more common in different parts of the world. Studies suggest that type 4 occurs more frequently in the Japanese and Korean populations than in people from other parts of the world [23]. Hermansky–Pudlak syndrome is the most common type of albinism in Puerto Rico, with a frequency of 1 case per 2,700 population. This disorder is very rare in other parts of the world. The first case of adult Hermansky–Pudlak was documented in Malaysia in 2012 [24].
(F)
Common to all types of OCA, there is reduced visual acuity and ocular nystagmus and these distinguish OCA from other forms of congenital diffuse hypopigmentation. The ocular abnormalities are due to misrouting of the optic nerve and this may result from deficiency of tyrosinase.
(G)
In routine clinical practice, babies with early diffuse hypopigmentation that involve skin, hair, and eyes should undergo a systemic evaluation to exclude other rarer disorders. For example, the presence of bleeding diatheses may be a sign of Hermansky–Pudlak syndrome and primary immune deficiency with recurrent infections points to Chediak–Higashi syndrome. If mental retardation with neurological regression is identified, blood and urine investigation is indicated to rule out phenylketonuria. Other inborn errors of metabolism, i.e., histidinemia and homocystinuria, can cause generalized diffuse hypopigmentation too (Table 3.6)
Table 3.6
Classification of disorder with cutaneous and ocular albinism
Disorder | Genetics | Pathogenesis | Clinical features |
|---|---|---|---|
OCA Type 1 A (Tyrosinase negative) | Autosomal recessive TYR gene on chromosome 11q14–q21 | Mutation in tyrosinase (TYR) gene with absent of tyrosinase activity As a result, melanosomes contain no melanin | 1. Skin and hair and eyes (a) Skin: white to pale pink (b) Hair: white (c) Eye: gray translucent iris with visual impairment and nystagmus No retinal pigment 2. Onset and progression (a) Onset at birth (b) No improvement with age. However, the hair may become yellowish later in later life |
OCA type 1B (Tyrosinase positive) | Autosomal recessive TYR gene on chromosome 11q14–q21 | Mutation in Tyrosinase (TYR) gene with variable reduction in tyrosinase activity Tyrosinase activity is temperature-sensitive | 1. Skin and hair and eyes (a) Skin: white to light pigmented (b) Hair: white to light yellow (c) Eye: gray translucent iris with visual impairment 2. Onset and progression [26] (a) Onset at birth and develop some pigmentations with age (b) Pigmented nevi and freckles are present |
OCA type 2 (Tyrosinase positive) | Autosomal recessive P gene on chromosome 15q11.2–q12 [27] | Mutation in P gene that involving in tyrosinase processing and transport [28] | 1. Skin, hair and eyes (a) Skin: creamy white to light brown (b) Hair: light yellow to light brown (c) Eye: blue, tan, hazel irides with visual impairment, nystagmus and minimal retinal pigment 2. Onset and progression (a) Onset at birth and darken with age (b) Freckles and pigmented nevus are seen |
OCA Type 3 (Rufous) | Autosomal recessive TYRP1 gene on chromosome 9q23 [29] | Mutation in TYRP1 gene that encodes dihydroxyindole carboxylic acid oxidase [30] | 1. Skin, hair, eyes (a) Skin: light brown to red bronze (b) Hair: light brown to red brown (c) Eyes: light brown with nystagmus 2. Onset and progression (a) Onset at birth and may darken with age (b) No freckles and nevus |
OCA type 4 | Autosomal recessive MATP gene on chromosome 5p13.3 | Mutation in MATP gene that encodes melanosomal protein [31] | 1. Skin, hair and eyes (a) Skin: creamy white to brown (b) Hair: silvery white to light yellow at birth (c) Eye: light brown with nystagmus 2. Onset and progression (a) Onset at birth and may darken with age. Freckles seen |
Hermansky–Pudlak syndrome | Autosomal recessive | HPS type 1–type 7 due to mutations of 7 different genes that encodes proteins involved in biogenesis of lysosome related proteins | 1. Skin, hair and eyes (a) Skin: variable from creamy white to almost normal skin (b) Hair: creamy to red brown (c) Eye: blue to brown irides with visual impairment and nystagmus 2. Onset and progression: onset at birth 3. Extracutaneous Increased in bleeding tendency, pulmonary fibrosis, granulomatous colitis, renal failure and cardiomyopathy due to ceroid deposition |
Chediak–Higashi syndrome | Autosomal recessive LYST gene on chromosome 1q42–1q43 | Mutation of LYST gene that controls lysosome trafficking Giant lysosomes seen in many cells including neutrophils, monocytes, hepatocytes and melanocytes | 1. Skin, hair and eyes (a) Skin: white skin (b) Hair: blond to silver (c) Eyes: blue to brown irides, strabismus and photophobia, minimal retinal pigment 2. Onset and progression (a) Onset at birth and may darken with age 3. Extracutaneous (a) Multiple infections and risk of lymphoma |
(H)
In terms of treatment, the aggressive sun protection to prevent actinic damage and early development of skin cancer is needed. Regular eye assessment is essential for their eye problems. Correction of refractive errors with spectacles or contact lenses improves visual acuity. Dark glasses may alleviate photophobia.
(I)
In terms of its psychosocial impact , albinism often has social ramifications because patients may get stigmatized as a result of the difference in appearance from their families, peers, and other members of their ethnic group. This is true especially among skin of color.
Group B-2: Early-Onset Generalized Diffuse Hypopigmentation with Skin and Hair Involvement
For children with early-onset generalized diffuse hypopigmentation, full eye assessment and follow-up is essential in the diagnostic process. This is because some genetic disorders, e.g., Griscelli, Elejalde, and Menkes syndromes (Table 3.7), result in pigmentary dilution of the skin and hair, but spare the eyes. These patients should also be screened for neurological, immunological, and other systemic abnormalities (Table 3.8). Other conditions that need to be considered are ectrodactyly–ectodermal dysplasia–cleft lip/palate (EEC) syndrome and selenium deficiency among patient on long-term total parenteral nutrition [25].
Table 3.7
Group B-2: early onset generalized diffuse hypopigmentation of skin and hair only
Disorder | Genetic | Pathogenesis | Clinical features |
|---|---|---|---|
Griscelli syndrome (GS) | Autosomal recessive 3 types identified (A) Type 1 GS [32]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|