Elizabeth Starkey, Imti Choonara, Helen Sammons • Understand the principles of pharmacokinetics in children • Know the major pathways of drug metabolism in children and how they vary with age • Know about some of the commoner adverse drug reactions that occur in children • Understand the mechanisms by which adverse drug reactions occur in children • Know the principles of safe prescribing • Be aware of the most frequent types of medication error • Understand the pharmacology of some of the commonly used drugs in children • Understand the importance of the rational use of medicines in children Understanding how the body handles different medicines is important for all clinicians when prescribing medications. For effective and safe prescribing, we need to make sure that we do not under-dose a medication, making it ineffective, but also that we do not give a dose that causes toxic effects. In paediatrics, significant physiological and developmental differences add to the challenges of safe prescribing. The key parameters of clinical pharmacology will be described below and the differences seen in the different paediatric age groups will be highlighted. Pharmacokinetics (PK) describes the course of a drug within the body; this is expressed as the dose given and concentrations in different parts of the body (usually plasma). It includes how it is absorbed, distributed, metabolized and finally excreted. These will each be discussed separately, along with the common equations used. Pharmacokinetics allows us to understand the profile of a drug’s concentration over time and recommend a drug dosing regimen or, when faced with a novel paediatric drug therapy, provides us with knowledge to prescribe safely and effectively. Mathematical formulae are available that describe the inter-relationship between clearance, volume of distribution and elimination half-life. If a drug is given intravenously, 100% of the dose will enter the blood stream, but for any other route less than 100% of the dose will be absorbed. This is because it must overcome chemical, physical, mechanical and biological barriers; the percentage that enters the systemic circulation is known as its bioavailability. Absorption is the process of drug movement from the site of administration or application into the systemic circulation. It is often reduced following oral administration in the neonatal period. Additionally, in the neonatal period, pH is elevated within the stomach. This increase in gastric pH affects the bioavailability of medicines. This higher gastric pH increases the absorption of weak base drugs such as penicillins and decreases the absorption of acidic drugs such as phenobarbital and phenytoin, which may therefore require a larger oral dose. Fortunately, in sick neonates most medicines are given intravenously and therefore absorption is not usually a clinical problem. For children, there are other developmental changes to drug absorption that occur in different systems. Intramuscular absorption depends on skeletal muscle blood flow; neonates have poor muscle bulk and poor muscle density, reducing bioavailability. Percutaneous absorption is enhanced in childhood due to the larger surface area of the skin relative to the body weight, and better skin hydration and perfusion. Young infants and neonates also have increased absorption due to their skin being thin. This increases systemic absorption and therefore potential side effects of topical medications. A historical catastrophic example of this is the topical disinfectant hexachlorophene in neonates, which caused neurotoxicity and death. Developmental changes in pulmonary structure and capacity in young patients may also alter the patterns of inhaled drug absorption (see also Chapter 17, Respiratory medicine). This is not a physiological volume, but rather an apparent volume into which the drug would have to distribute to achieve the measured concentration. The volume of distribution is usually defined in litres or litres/kg. It is calculated by dividing the amount of drug by the plasma concentration: A small volume of distribution indicates a drug is largely retained within the systemic circulation, whereas a large volume of distribution means a drug is well distributed into other peripheral compartments. Water-soluble drugs, such as gentamicin, therefore have a volume of distribution that is similar to the extracellular fluid volume. Drugs that are highly bound to plasma proteins, such as phenytoin, have a low volume of distribution. Differences between paediatric and adult patients stem mainly from the fact that neonates and young children have a higher proportion of body water and lower concentrations of plasma proteins. Knowing the volume of distribution of a drug is useful when determining what loading dose is to be given. This is calculated from the following formula: For example, in order to achieve a peak gentamicin concentration of 10 mg/L in a neonate weighing 1 kg, where the volume of distribution is known to be 0.5 L/kg, one would multiply 10 mg/L by 0.5 L/kg by 1 kg. This equates to 5 mg. If some of the drug is already present in the patient, one can subtract the measured plasma concentration from the target concentration in order to calculate the dose that is required. Total body clearance is the ability of the body to remove a drug from the plasma or blood and is the sum of drug clearances of each organ. For many drugs, this is equal to hepatic clearance plus renal clearance. Renal clearance is determined by the clearance of an unchanged drug in the urine, whereas liver clearance can occur via biotransformation to a metabolite, which is subsequently excreted via the urine, and/or excretion of the unchanged drug into the biliary tract. It is defined as the volume (usually of plasma) that is completely cleared of drug in a given time period. In adults, clearance is therefore described in relation to volume/time (L/hour). In paediatric patients, clearance is also described in relation to body weight (either as L/hour/kg or mL/min/kg). Clearance can be used in conjunction with the target steady state concentration (CSS) to calculate the rate of administration of a drug given intravenously. This is shown in the following equation: Or for children, where a dose and clearance are expressed in relation to body weight: This formula is appropriate for the administration of drugs given intravenously. For example, the maintenance dose of an intravenous aminophylline infusion to achieve a theophylline level of 10 mg/L in a 20 kg child where clearance is 0.087 mg/kg/hour is 10 mg/L × (0.087 × 20) = 17.4 mg/hour. For drugs that are given orally, one needs to take account of the bioavailability as well as the dosage interval between different doses. If a drug is given via regular bolus intervals, the ‘average’ target CSS is used as steady state fluctuates between the peak and trough and the dosing interval (π) is also added into the equation. The maintenance dose can therefore be calculated by the following formula: Maturation of renal function occurs during childhood. The maturation process of kidney structure and function is associated with prolongation and maturation of the tubules, increase in renal blood flow, and improvement of filtration efficiency. This knowledge allows us to provide a rational dose schedule for drugs exclusively eliminated via the kidneys. In general, the neonate will need longer dose intervals than the infant to maintain target concentrations. For example, the dose of benzylpenicillin changes from 25–50 mg/kg 12 hourly in a neonate <7 days to 8 hourly in a 7–28-day neonate and 4–6 hourly in children over a month old. Half life is a secondary pharmacokinetic parameter and is the time taken for the drug concentration (usually in the plasma) to decrease by half. Therefore, 50% of the dose will be eliminated in one half-life. It is inversely related to the clearance and can be calculated using a drug’s volume of distribution and clearance with the following equation: (Note: the value 0.693 is the natural logarithm of 2) Half-life can be used to determine the time it takes to achieve steady state and the time for a drug to be completely eliminated. It takes around 3–5 times the drug’s half-life to reach steady state and the same for it to be completely eliminated in constant dosing. Five half-lives is the time required for 97% of the drug to be eliminated (Fig. 36.1). For instance, the t1/2 of intravenous midazolam is of the order of 1.1 hours in 3–10-year-olds, therefore it takes around 3.3 to 5.5 hours to reach steady state. The half-life helps the clinician to establish an appropriate drug dosing interval. When a medication is given every half-life, the plasma concentration will have a twofold fluctuation over the dosing interval (see Fig. 36.1). For drugs with a half-life <6 hours, it is sometimes not practical to give frequent doses, so sustained release formulations are given (e.g. theophylline). In drugs with a very long half-life (e.g. amiodarone), a daily treatment may be appropriate. A loading dose helps to reach the steady state more quickly. The t1/2 of phenobarbital in neonates is 67–99 hours, so without a loading dose it could take 8–20 days to reach a steady state. Although drug half-lives are quoted in the literature, they represent average values mainly in adults and should be used cautiously. The pharmacokinetic principles outlined above assume that the drug follows first-order or linear pharmacokinetic characteristics. This means that the steady-state concentration changes in direct proportion to a drug dose alteration. However, for some drugs, the relationship is more complex. For example, phenytoin saturates the metabolizing enzymes at clinical doses. Subsequent increases in dosing cause a disproportionate elevation of the steady-state concentration. This is known as zero order or saturated kinetics.
Pharmacology and therapeutics
Introduction
Pharmacokinetics
Absorption
Volume of distribution (Vd)

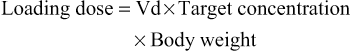
Clearance
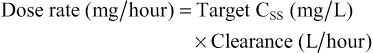
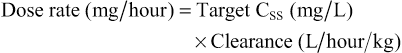
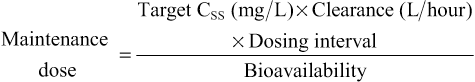
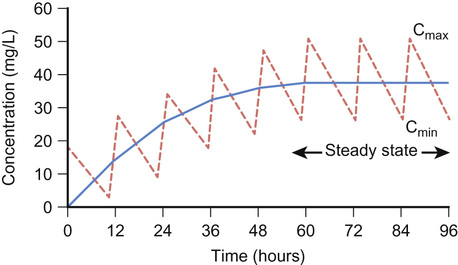
Half-life
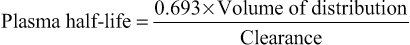
Drug–food interactions
Interactions between food and drugs can unintentionally reduce or increase the effect of an oral medicine, resulting in potential therapeutic failure or toxicity. Firstly, food intake also impacts on drug absorption by stimulating gastrointestinal secretions, pancreatic hormones, and bile salts (which lower gastric pH), as well as delaying stomach emptying and increasing gastrointestinal transit time. The size and content of a meal, especially those with a high fat content, also play a role and can reduce a drug’s rate of absorption.
Secondly, food has the ability to affect a drug’s bioavailability by interaction with the food constituents. A good example is the reduction in bioavailability of tetracyclines following dietary calcium caused by chelation. Food–drug interactions affecting metabolism, distribution or elimination are not very common, apart from interactions with grapefruit juice. Grapefruit juice contains potent inhibitors of the cytochrome P450. CYP3A4, a P450 enzyme, may markedly increase the bioavailability of drugs that it metabolizes, including ciclosporin, midazolam and carbamazepine.
Therapeutic drug monitoring
Therapeutic drug monitoring (TDM) consists of measuring plasma concentrations of the drug in order to improve its efficacy whilst reducing its toxicity (Box 36.1 outlines criteria for use). TDM is recommended for certain antibiotics, including the aminoglycosides and glycopeptides, in order to reduce potential toxicity. It may be beneficial in patients with poorly controlled epilepsy who are receiving carbamazepine, phenytoin or phenobarbital. Interpretation of the plasma concentration of a drug requires details of the time of administration of the drug and time of collection of the blood sample, as well as an understanding of why TDM has been requested.
Drug levels should only be taken once the drug has reached its steady state, unless there are concerns regarding toxicity. In general, trough levels measured just prior to drug administration provide accurate interpretation of drug concentrations. Peak levels are less accurate due to individual variability and are reserved for treatments with short half-lives where peak levels are associated with efficacy or toxicity, e.g. gentamicin.
TDM for aminoglycosides
This class of drug is bactericidal and works by irreversibly binding the 30S subunit of the bacterial ribosome, and interfering with bacterial protein synthesis. Aminoglycosides are mainly used for the treatment of severe Gram-negative infections, with tobramycin and gentamicin having some activity against Pseudomonas infections. Tobramycin is used frequently in children with cystic fibrosis. Gentamicin also works synergistically with β-lactams for the treatment of Gram-positive Staphylococci infections. This is why benzylpenicillin and gentamicin are used in combination for the treatment of group B streptococcal neonatal infections.
TDM is essential when using aminoglycosides because of the significant oto- and nephrotoxicity that can occur with these agents. It is thought that toxicity is associated with high trough concentrations. Studies in adults suggest that ototoxicity is more frequent than nephrotoxicity. Ototoxicity has been described following single doses and is thought to occur in 5–10% of adults who receive aminoglycosides. Both prolonged and repeated courses are thought to be risk factors for toxicity. Aminoglycosides used to be given three times daily but current practice is to give them once daily. This larger daily dose produces a higher peak level than the standard regime, which in turn increases the rate and extent of bacterial cell death. It also lengthens the post-antibiotic effect (suppression of bacterial regrowth) without increasing the risk of any drug toxicity. When multiple daily dose regimens are used, as well as a pre-dose (trough concentration), one should measure a one-hour (peak) post-dose concentration. Most hospitals in the UK provide a clinical pharmacy service to help interpret the plasma concentration and give advice regarding dose adjustment. The beneficial effect of discussing management with a clinical pharmacist has been demonstrated. In addition to minimizing toxicity, one needs to ensure that the individual patient receives a dose that is effective in treating the significant bacterial infection that the patient is likely to be suffering from.
TDM for glycopeptides
Glycopeptides are another group of antibiotics that require therapeutic drug monitoring and include vancomycin and teicoplanin. They act by interfering with the bacterial cell wall synthesis in Gram-positive bacteria. They bind to the end of the pentapeptide chains that are part of the growing cell wall structure. This inhibits the transglycosylation reaction and prevents incorporation.
Vancomycin and teicoplanin are used in intravenous form for the treatment of serious infections caused by Gram-positive cocci such as Staphylococcus aureus and coagulase-negative Staphylococcus. Vancomycin is the main treatment for patients with MRSA infections. It can also be given orally for the treatment of pseudomembranous colitis in the colon, usually caused by Clostridium difficile, which is rarely seen in children.
Like aminoglycosides, glycopeptides are nephro- and ototoxic and hence require TDM (Box 36.2). Variations in protocol occur throughout different hospitals about how to monitor and adjust vancomycin dosing, and local policies should be followed. Teicoplanin is less toxic than vancomycin but still requires monitoring. Adverse drug reactions due to vancomycin include red man syndrome, characterized by flushing and erythematous skin usually of the upper body and face. This is caused by a non-specific mast cell degranulation and can be avoided with a slow infusion rate.
Question 36.4
Drug metabolism
The following (A–H) is a list of drug metabolism pathways:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


