Phagocytic Disorder
INTRODUCTION
Phagocytosis is the process by which leukocytes internalize particles larger than 1 μm, such as microbes, apoptotic cells, and chemical substances.1 It is an essential component of the innate and adaptive immune response, as well as tissue remodeling and repair.2 Neutrophils, macrophages, and dendritic cells are professional phagocytes that are derived from the common myeloid progenitor.3 As the invading microbes cross the skin and mucosal barrier and enter the tissue, they are recognized by resident macrophages and dendritic cells through germline-encoded pattern recognition receptors. The release of proinflammatory cytokines activates local endothelial and epithelial cells, which express chemoattractants and adhesion molecules. Circulating neutrophils adhere to the endothelium and migrate through the endothelial lining into the tissues.4,5
Phagocytosis is a receptor-mediated process. Apart from pattern recognition receptors, foreign particles bound by complements and immunoglobulins in the circulation and interstitial fluid are recognized by phagocytes through the opsonic receptors.6,7 The ingested microorganism is degraded in the phagolysosome, and the presentation of antigenic peptide on the surface of the phagocytes leads to lymphocyte activation. The mechanism of killing is accomplished by de novo synthesis of reactive oxygen species and release of proteases stored in lysosomal granules into the phagolysosome.
Phagocytosis is the first line of defense against infection and forms the link between innate and adaptive immunity.8 Phagocytic defects commonly present in the neonatal period and early infancy. The spectrum of disorders includes numerical deficiency and functional defects such as impaired adhesion, chemotaxis, ingestion, degranulation, and intracellular killing.
PATHOPHYSIOLOGY
Neutrophil Count in the Neonates
The absolute neutrophil count (ANC) is calculated by multiplying the total white blood cell count (WCC) by the percentage of bands and mature neutrophils. The ANC of the newborn varies with gestational age, postnatal age, birth weight, geographical factors such as altitude, and clinical factors, including intrapartum oxytocin administration and maternal hypertension.9–11 The commonly used ANC reference range in term and near-term neonates was established in 1979 by Manroe et al.12 At birth, the ANC ranges from 1800 to 5500/mm3, which subsequently increases 3- to 5-fold and peaks at 12–18 hours of life. Gradual fall in ANC occurs by 24 hours, ranging from 1800 to 7200/mm3 at 60 hours and remaining at 1800 to 5400/mm3 within the first 28 days. The ANC reference range in very low birth weight (VLBW) neonates was published in 1994 by Mouzinho et al.13 In their study of 193 premature infants born at mean gestational age of 29.5 weeks with mean birth weight of 1157 g, the lower boundary of ANC was found to be 500/mm3 at birth, rose less steeply to 2200/mm3 at 18–20 hours, then decreased to 1100/mm3 at 60 hours. From 60 hours to 28 days of life, the normal range of ANC is 1100–6000/mm3.
Subsequent studies showed that both neutropenia and neutrophilia occur more commonly in premature neonates compared with those born near or at term, reflecting their immature granulopoietic regulation.14 The reported prevalence of neutropenia in preterm infants varies from 6% to 58%, depending on the reference range used and the specific population risk factors.15–18 Rebound neutrophilia is commonly observed in the second week of life in the absence of infections. Idiopathic neutropenia of prematurity refers to neutropenia that occurs after 3 weeks of life. It was reported in 26% of preterm neonates whose birth weights were appropriate for gestational age (AGA) and generally resolves spontaneously without complications.19–21
Neutropenia
Neutropenia is the absolute reduction in the number of neutrophils in the circulation. It is usually defined as an ANC of less than 1500/mm2, but in neonates factors such as age and maturity, as mentioned, should be taken into account. The peripheral neutrophil count reflects the equilibrium between the intra- and extramedullary compartments. Neutropenia can be caused by (1) decreased production; (2) increased consumption or destruction; (3) excessive margination; (4) excessive apoptosis, or a combination of these causes. Acquired or secondary neutropenia are much more common than congenital neutropenia (Table 51-1). Frequently identified causes include maternal hypertension, sepsis, immune-mediated destruction, and drug exposure.
Table 51-1 Acquired or Secondary Causes of Neutropenia in Neonates
Maternal or Materno-Fetal Factors
Maternal hypertension
Maternal autoimmune neutropenia
Neonatal alloimmune neutropenia
Postnatal Factors
Birth asphyxia
Bacterial sepsis
Viral infections
Drug-induced agranulocytosis
Autoimmune neutropenia
Hypersplenism
Nutritional deficiency
Maternal preeclampsia, birth asphyxia, and sepsis are common perinatal events associated with neutropenia presenting in the first few days of life. It was reported that up to half of premature infants born to hypertensive mothers are neutropenic, attributable to a placenta-derived granulopoiesis inhibitor.22,23 Association of neutropenia and early-onset sepsis (<48 hours) caused by group B streptococcus and other bacterial infections has been well demonstrated.24–26 Circulating neutrophils and platelets are diminished as a result of depleted reserve in the bone marrow. It is important to realize the significance of neutropenia and potential sepsis within the first day of life, when the ANC should normally be rising to its peak. Viral infections, such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and parvovirus, can lead to bone marrow suppression and neutropenia, which may be accompanied by anemia or thrombocytopenia.
Immune-mediated neutrophil destruction can be caused by transplacental passage of maternal immunoglobulin (Ig) G antineutrophil antibodies in maternal autoimmune neutropenia and neonatal alloimmune neutropenia.27 Neonatal alloimmune neutropenia is mediated by maternal antibodies against fetal neutrophil antigens inherited from the father, analogous to hemolytic disease of the newborn (HDN) caused by rhesus isoimmunization. Unlike HDN, alloimmune neutropenia can occur in the firstborn child.28 Sensitization occurs when fetal neutrophils enter the maternal circulation during pregnancy, and antibody-mediated neutrophil destruction occurs in utero and after delivery. In most cases, maternal alloantibodies are directed toward neutrophil-specific antigens, human neutrophil antigen (HNA) 1a and HNA-2a, and less commonly HNA-1b or others.29 Antibodies against human leukocyte antigens (HLA) may also cause alloimmune neutropenia.30 The incidence is estimated to be 1 in 1000 newborns.31 Affected infants are neutropenic at birth. Most of them are asymptomatic; some may present with omphalitis, delayed separation of the umbilical cord, mild skin infections, fever, or pneumonia, but severe infections are rare.32,33
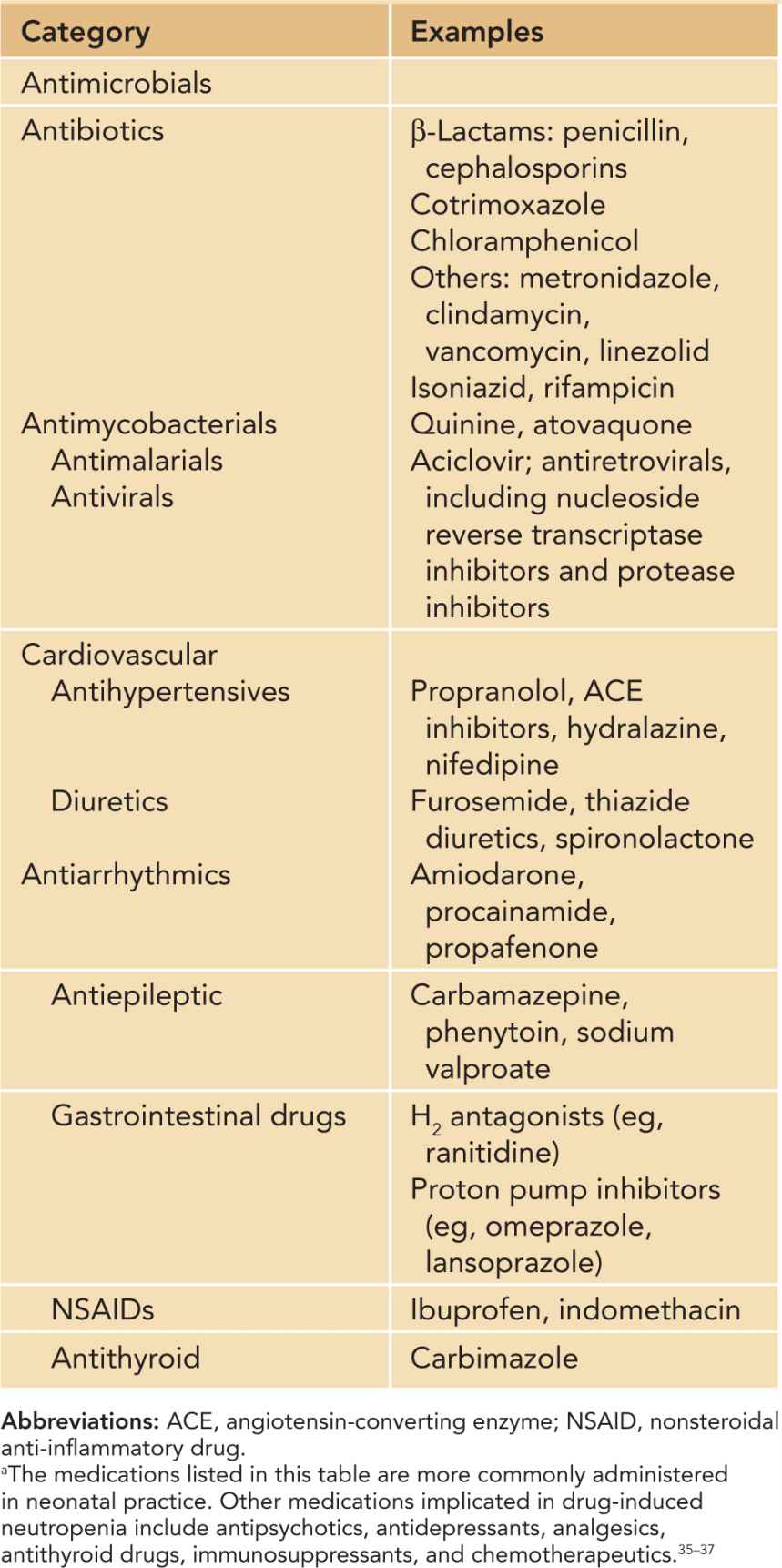
Idiosyncratic drug-induced neutropenia is uncommon in neonates and children. The overall population incidence is 2.4–15.4 per million per year.34 The incidence increases with age, and only 10% of cases occur in children and young adults. Among the long list of medications associated with neutropenia,35–37 drugs that are more commonly encountered in neonatal practice include antimicrobials, antiepileptic agents, antithyroid drugs, and cardiovascular and some gastrointestinal medications (Table 51-2). Neutropenia is usually profound, often below 200/mm3. Neutropenia associated with carbimazole, propylthiouracil, phenytoin, hydralazine, and procainamide is immune mediated, whereas sulfonamides, trimethoprim-sulfamathoxazole, carbamazepine, sodium valproate, and antivirals such as ganciclovir cause neutropenia by suppressing granulopoiesis. Idiosyncractic neutropenia caused by penicillin is mediated by both mechanisms.37
Table 51-2 Idiosyncratic Drug-Induced Neutropeniaa
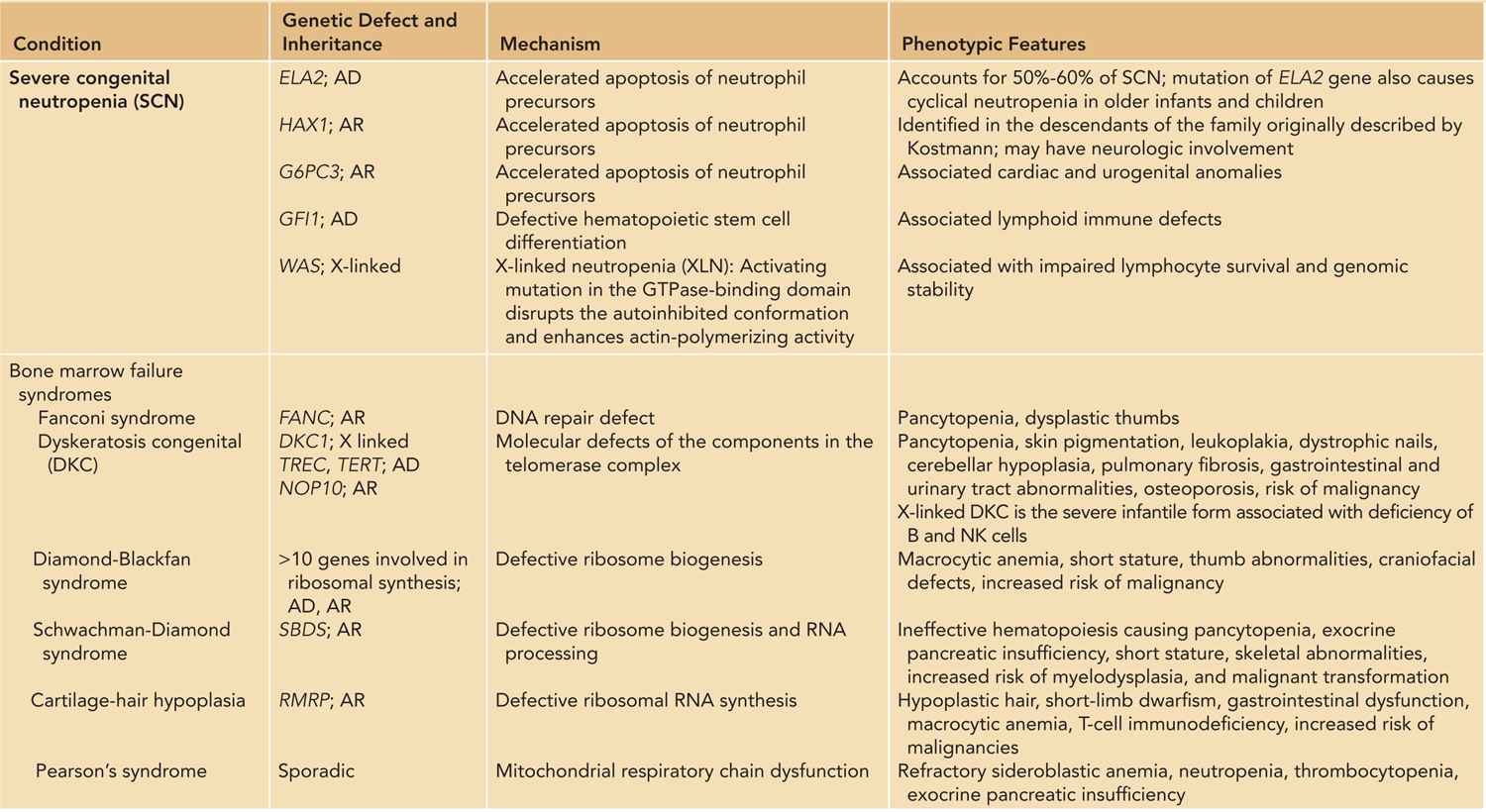
Neutropenia with underlying genetic defects are rare. They include severe chronic neutropenia (SCN), bone marrow failure syndromes, metabolic disorders, and neutropenia associated with cellular and humoral immunodeficiencies (Table 51-3).38–42 SCN involves a genetically heterogeneous group of disorders characterized by maturation arrest of myelopoiesis at the level of the promyelocyte/myelocyte stage in the bone marrow, resulting in profound neutropenia.43 Mutation of the neutrophil elastase 2 (ELA2) gene inherited in an autosomal dominant manner accounts for 50%–60% of patients with SCN. Mutations of other genes, such as GFI1, HAX1, SBDS, WAS, and G6PC3, altogether account for less than 5% of SCN, so the genetic basis in 40% of cases remains unknown.44 While loss-of-function mutation in the WAS gene causes classical Wiskott-Aldrich syndrome or X-linked thrombocytopenia, mutations in the guanosine triphosphatase (GTPase)–binding domain of WAS abrogates the autoinhibitory conformation and produces a constitutively activated form, resulting in enhanced actin polymerization and cellular apoptosis.45
Table 51-3 Neutropenia with Single-Gene Defects: Inheritance, Pathogenetic Mechanisms, and Clinical Features
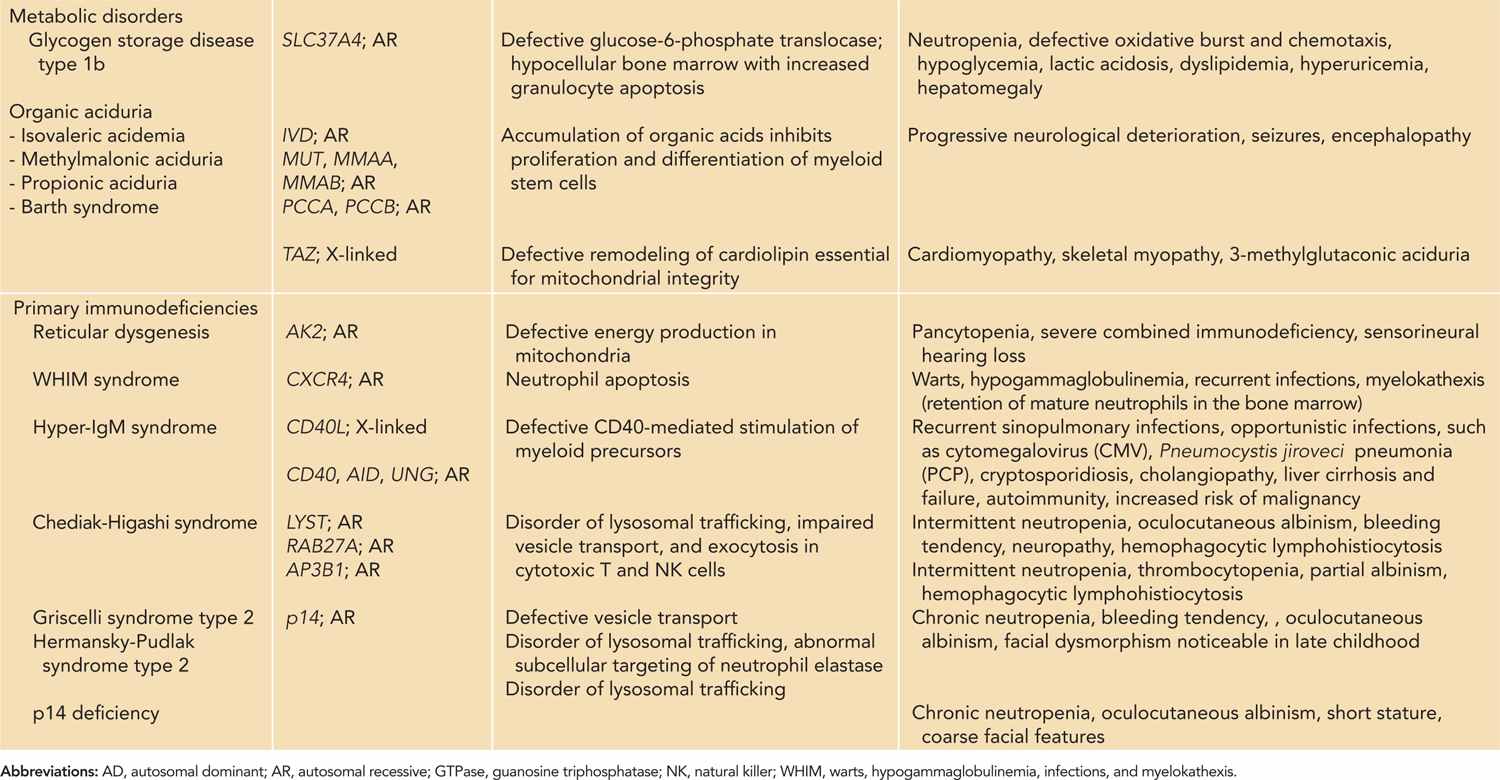
As neutrophils have a short half-life with high cellular turnover, they are most susceptible to derangements in cellular metabolism. The pathogenesis of congenital bone marrow failure syndromes involves various basic cellular processes, including ribosome biogenesis, mitochondrial metabolism, and DNA repair.46,47 Infants with congenital bone marrow failure syndromes often have pancytopenia. Skeletal malformations are common, and some are associated with lymphocyte abnormalities and increased risk of malignancy. Neutropenia also occurs in metabolic disorders such as glycogen storage type 1b48,49 and organic aciduria.50
Some forms of humoral and cellular immunodeficiencies are associated with neutropenia,51 such as reticular dysgenesis52,53 and hyper-IgM syndrome.54–56 Disorders of abnormal lysosomal trafficking such as Chediak-Higashi syndrome, Griscelli syndrome type 2, and Hermansky-Pudlak syndrome type 2 are characterized by partial oculocutaneous albinism, bleeding tendency, neutropenia, and hemophagocytic lymphohistiocytosis.57,58 Immune cytopenia occurs in autoimmune lymphoproliferative syndrome (ALPS) and is discussed in Chapter 60. Other forms of primary immunodeficiencies associated with neutropenia include humoral deficiencies that include X-linked agammaglobulinemia,59,60 common variable immunodeficiency,61,62 and WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis),63 but they do not present in the neonatal period.
Susceptibility to infections depends on the ANC, adequacy of neutrophil reserve pool in the bone marrow, and other factors, such as integrity of mucosal barriers and coexisting cellular immune deficiencies. Patients with ANC below 500/mm3 or neutropenia secondary to chemotherapy, marrow failure, or marrow exhaustion are at the greatest risk of overwhelming bacterial and fungal infections.64
Disorders of Phagocyte Function
Defects of Neutrophil Adhesion and Chemotaxis
To reach the focus of infection in the extravascular space, circulating neutrophils must leave the bloodstream by migrating through the vascular endothelium. Tissue trauma or inflammation leads to activation of endothelial cells, which express adhesion molecules such as selectins and integrins.65 Selectin binding brings leukocytes into contact with the endothelium, on which leukocyte rolling is followed by integrin-mediated adhesion and transendothelial migration via paracellular or transcellular paths to the site of inflammation guided by chemotactic factors.66
At present, 3 forms of leukocyte adhesion deficiency (LAD) are known, and all are autosomal recessive disorders.67 LAD type I is caused by a defect of the common chain (CD18) of β2 integrins, which serve as receptors for the opsonic complement fragment C3b and intercellular adhesion molecules 1 and 2 (ICAM-1 and ICAM-2, respectively) expressed on endothelial cells and leukocytes. The β2-integrin molecule is a heterodimer of α chains (CD11a, CD11b, or CD11c) and β chain (CD18). Defective heterodimer formation leads to profound impairment of leukocyte emigration from the bloodstream to the site of inflammation in the extravascular space. As CD11b/CD18 is the predominant receptor for the complement fragment C3bi, ingestion of C3bi-opsonized microbes and C3bi-mediated phagocytic activities is also affected.68
Patients with LAD exhibit marked leukocytosis, and the ANC may reach up to 100,000/mm3 during acute infections. The lack of pus formation at the site of inflammation despite persistent granulocytosis is characteristic. Impaired local control of infection predisposes to systemic spread. Bacterial infections and skin abscesses are often apparent in the early neonatal period. Delayed separation of the umbilical cord and omphalitis with persistent discharge are pathognomic of LAD. Common causative organisms include Staphylococcus aureus and gram-negative enteric bacteria. Fungal infections are also common. Infant and children with LAD often have severe gingivitis and periodontitis. In addition, healing of traumatic or surgical wound is significantly impaired.69 Clinical severity correlates with the residual surface expression of CD18.70
Leukocyte adhesion deficiency type II is a disorder of fucose metabolism caused by defective guanosine diphosphate (GDP)–fucose transporter.71 Neutrophils fail to roll on the vascular endothelium because of the absence of SLeX, a fucosylated ligand for selectins expressed on the surface of leukocytes. Infective and inflammatory manifestations are similar to LAD I, but additional features in LAD II include coarse facial features, short stature, moderate-to-severe mental retardation, and absence of H antigen on erythrocytes (Bombay phenotype). LAD II is extremely rare and has been reported in fewer than 10 patients in the literature.72
Leukocyte adhesion deficiency type III is caused by defect of Kindlin-3, a hematopoietic structural protein that mediates integrin activation.73 In addition to leukocyte adhesion defect, platelet aggregation is abnormal and causes severe bleeding tendency.74,75 Other features of LAD III include increased bone density resembling osteopetrosis and neurodevelopmental disabilities.76
Defects of Phagocyte Killing
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is a disorder of phagocytic oxidative burst as a result of impaired nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity.77 The NADPH oxidase is a multimeric enzyme complex that catalyzes the transfer of electrons from cytoplasmic NADPH to molecular oxygen. The membrane-bound flavocytochrome b558 of NADPH oxidase consists of gp91phox and p22phox.78 Phagocytosis of microorganisms triggers the translocation of 4 cytosolic factors (p47phox, p67phox, p40phox, and Rac2) to the cell membrane, where they associate with gp91phox and p22phox (Figure 51-1). Electrons transported from cytoplasmic NADPH via the activated NADPH oxidase convert molecular oxygen to superoxide anion in the phagosome, generating bactericidal oxidants such as hydrogen peroxide and hypohalous acid. At the same time, the activation of antimicrobial proteases and release of neutrophil extracellular traps (NETs) further augments antimicrobial defense. Granular proteases such as neutrophil elastase and cathepsin G sequestered in the azurophilic granules enhance killing within the phagolysosomes,79 and NETs entrap bacteria and fungi and facilitates extracellular killing.80
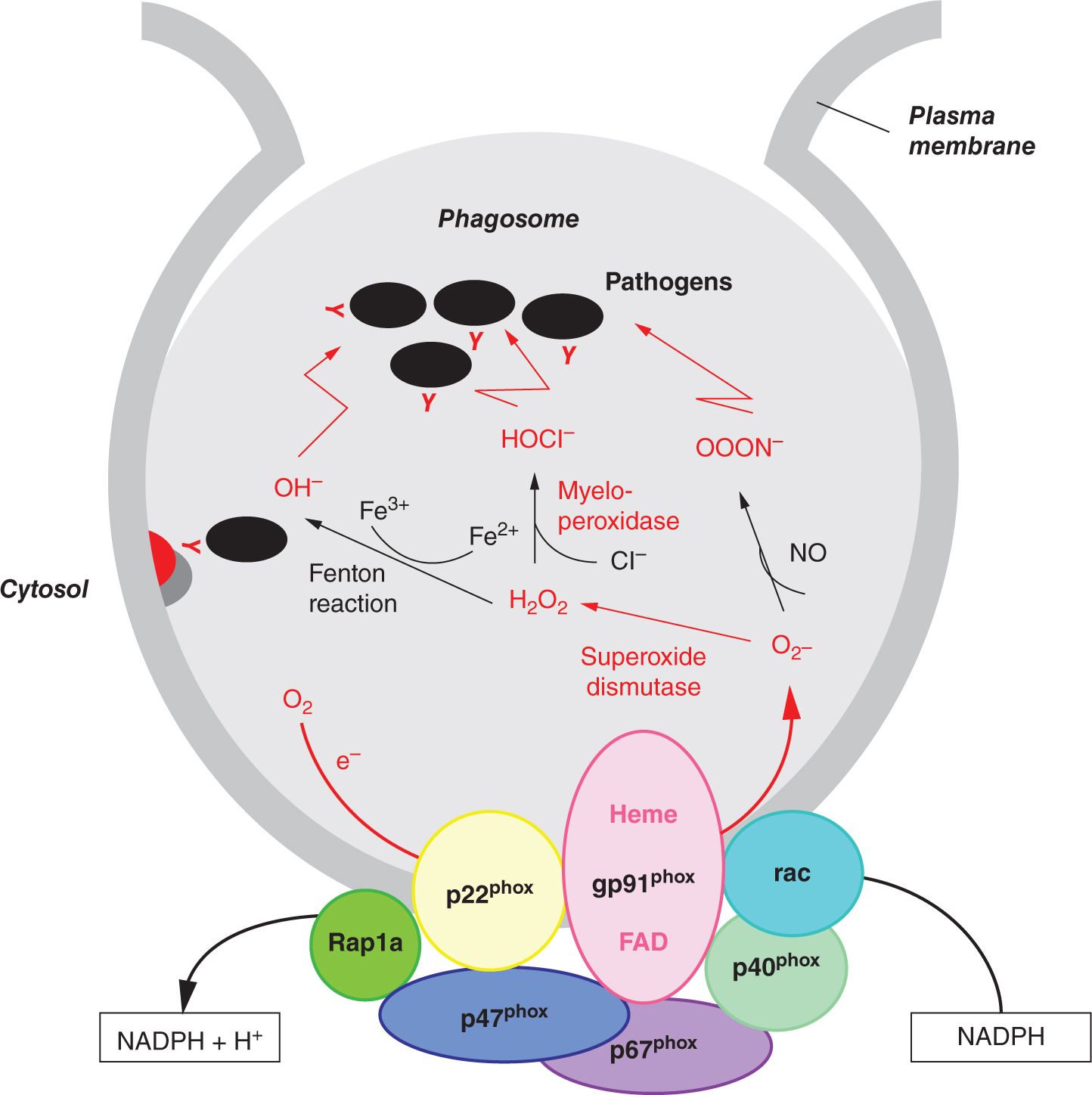
FIGURE 50-1 Components of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and respiratory burst.
Molecular defects of various components of the NADPH oxidase complex have been identified to cause CGD. X-linked mutations in the CYBB gene encoding gp91phox account for 60%–70% of CGD.81 Genetic defects of p22phox (CYBA), p47phox (NCF1), p67phox (NCF2), and p40phox (NCF4) are inherited in an autosomal recessive manner.82
Chronic granulomatous disease affects 1 in 200,000 to 250,000 individuals. Most patients present in early infancy, but CGD with attenuated phenotypes is increasingly diagnosed in adulthood. Patients with CGD are susceptible to S. aureus; Nocardia species; gram-negative bacteria such as Salmonella species, Serratia marcescens, and Burkholderia cepacia; fungal infections such as Aspergillus; and mycobacteria, including M. tuberculosis and M. bovis BCG (bacillus Calmette-Guérin.83 Common presentations in the neonatal period and early infancy include recurrent skin and perianal abscesses, suppurative adenitis, and pneumonia. Bacterial and fungal dissemination by the hematogenous route may lead to septicemia, liver abscess, osteomyelitis, meningitis, and brain abscess. Infants often have a history of prolonged antibiotic use as well as multiple incision and drainage procedures for recurrent abscesses. Those who receive BCG vaccine at birth may present with ulceration at the site of inoculation and enlargement of ipsilateral axillary lymph nodes, and some may develop systemic dissemination.84,85
Patients with CGD have a unique susceptibility to Chromobacterium violaceum, which is a soil and water saprophyte mostly confined to tropical and subtropical areas.86 Chromobacterium violaceum infection was first reported to occur at unusually high frequency among patients with CGD living in the southeastern United States, such as in Florida and Louisiana.87
Children and adults with CGD may have hyperinflammatory manifestations such as colitis, granuloma, and excessive inflammatory reaction at the site of trauma.88 The mechanisms of exaggerated inflammatory response in CGD include reduced degradation of phagocytosed materials causing persistent phagocytic activation, induction of Th17 cells, impaired neutrophil apoptosis, dysregulation of the redox-sensitive anti-inflammatory molecule Nrf2, and activation of NLRP3 inflammasome.88,89 Hyperinflammatory lesions may affect any part of the gastrointestinal tract, from the oral cavity to anus. Granulomatous colitis can mimic Crohn disease and manifests as bloody diarrhea, malabsorption, anemia, and failure to thrive.90 Granuloma may obstruct hollow viscera such as the gastric outlet and urethral orifices.
Myeloperoxidase Deficiency
Myeloperoxidase is a component of the azurophilic granules in phagocytes. It catalyzes the reaction of hydrogen peroxide with chloride ion to form hypochlorous acid, which is then converted to chlorine.79 As superoxide is generated normally, intracellular killing of pathogens is only minimally affected, except impaired candida killing, which can be demonstrated by in vitro assays. In general, individuals with myeloperoxidase deficiency are asymptomatic, but those who have concomitant diabetes mellitus are susceptible to severe candidiasis.91,92
DIFFERENTIAL DIAGNOSIS
The main clinical presentations of phagocytic disorders in the neonatal period are infections and abnormal wound healing. Sepsis can be the cause or result of neutropenia and should be treated aggressively with broad-spectrum antimicrobial therapy. Neutrophil count should be interpreted with caution and serially monitored, and factors including gestational age, postnatal age, perinatal events, and drug exposure should be taken into account. Secondary causes of neutropenia are much more common than inherited defects of myelopoiesis and syndromal disorders but should be considered if neutropenia is severe and persistent, when other cell lineages are involved, and in the presence of coexisting congenital anomalies. Studies of phagocytic functions are warranted if clinical features are suggestive and neutrophil count is normal.
Family History
Inherited neutrophil disorders can be suggested by family history, such as recurrent infections and unexplained early infant deaths. Family history of hematological malignancy may be elicited in patients with bone marrow failure syndromes or severe congenital neutropenia. The pattern of inheritance may be apparent by obtaining a detailed family pedigree. X-linked phagocytic disorders include CGD caused by gp91phox defect, X-linked dyskeratosis congenita, X-linked neutropenia arising from an activating mutation in WAS, and CD40 ligand deficiency. Parental consanguinity increases the chance of autosomal recessive disorders. Infants with neonatal lupus syndrome can have severe neutropenia, and relevant maternal history should be sought.93,94 Maternal full blood cell count should be reviewed to look for neutropenia.
Physical Examination
On physical examination, the clinician should aim to identify the sites of infection and features suggestive of syndromal disorders. The oral cavity should be inspected for candidiasis. In older infants, gingivostomatitis may be apparent. Cutaneous pyogenic infections include impetigo, cellulitis, and abscess. The perianal region should be examined for induration, abscess, fistula formation, and scars. There may be persistent inflammation and discharge from the umbilicus or features suggestive of abnormal healing. Clinical signs of invasive infections, such as meningitis, osteomyelitis, and deep organ abscess should be sought. Failure to thrive is common in infants with chronic recurrent infections. Skeletal anomalies and growth retardation are often identified in patients with congenital bone marrow failure syndromes, such as dysplastic thumbs in Fanconi anemia and short-limb dwarfism in cartilage-hair hypoplasia. Cutaneous hypopigmentation is a feature common to various disorders of lysosomal trafficking, such as Chediak-Higashi syndrome and Griscelli syndrome type 2.
Recurrent Skin Abscess
Neutropenia and functional phagocytic disorders are the most important causes for recurrent skin abscess in infancy. Infants with eczema and staphylococcal carriage may also develop recurrent cellulitis and cutaneous abscesses, but invasive disease is unlikely. Diabetes mellitus impairs phagocytic function and predisposes to skin infections, but neonatal diabetes mellitus is a rare condition and primarily presents with intrauterine growth retardation, hyperglycemia, and dehydration. Humoral and complement deficiencies also increase susceptibility to pyogenic infections. Other primary immunodeficiency disorders associated with pyogenic skin infections include humoral deficiencies, complement defects, and hyper-IgE syndrome.95
Hyper-IgE syndrome is characterized by eczematous eruption, cutaneous cold abscesses, recurrent pneumonia and pneumatocele, pneumothorax, and elevated serum IgE.96,97 Infants with autosomal dominant hyper-IgE syndrome (Job syndrome) characteristically present with erythematous skin rash in the neonatal period98,99 and later develop somatic features, including coarse facies, scoliosis, bone fractures, and abnormal dentition. Patients with autosomal recessive hyper-IgE syndrome have additional susceptibility to viral infections, including molluscum contagiosum and warts.100,101 T-cell immunodeficiency should always be considered and ruled out in young infants with recurrent infections, and human immunodeficiency virus (HIV) testing should be performed as appropriate.
Perianal Abscess and Fistula
Perianal sepsis is not an uncommon condition in young infants and has a male preponderance. Most initially present with perianal abscess, subsequently progress to or recur as fistula-in-ano. The etiology appears to be related to congenital anomalies of anal crypts that predispose to abscess and fistula formation. In most healthy children, long-term or serious complications from perianal abscess or fistula are rare. Recurrence of perianal abscess is likely if a coexisting fistula is not identified and resected at the time of primary drainage.102,103 Phagocytic disorders should be considered if there is frequent recurrence and poor healing. Persistent perianal fistula and diarrhea are also presenting symptoms of neonatal/infantile-onset inflammatory bowel disease, which was recently identified as a group of genetic disorders in interleukin (IL) 10 signaling.104,105
Omphalitis and Delayed Separation of the Umbilical Cord
Separation of the umbilical cord normally occurs between 5 and 8 days after birth. The incidence of omphalitis in developed countries is estimated to be 0.5%–1.0%.106 Omphalitis delays the process of umbilical vessel obliteration and hence cord separation. Common risk factors for omphalitis include nonsterile delivery, maternal genital tract infection, prolonged rupture of membrane, prematurity, low birth weight, procedural complications such as umbilical cord catheterization, and improper handling and care of the umbilical cord stump.107,108 Omphalitis commonly occurs in neonates with neutropenia and phagocytic disorders, and delayed separation of the umbilical cord is characteristic of LAD.
BCG Complications
BCG vaccine complications such as ulceration, persistent discharge, and scarring are well recognized in infants who receive BCG vaccine in the neonatal period. Some of them may develop abscess at the site of inoculation, and the incidence of regional lymphadenitis is estimated to be 1 in 2800 vaccinations.109 Occurrence of BCG-related complications depends on external factors such as vaccine strains and technique of administration, and most affected infants are otherwise healthy.110–112 However, distant spread and systemic dissemination raise the alert for primary immunodeficiencies.113–117 T-cell deficiency and HIV infection should be excluded in such conditions. BCG complications also occur in genetic defects of the IL12–interferon-γ axis, collectively known as Mendelian susceptibility to mycobacterial diseases (MSMD).118,119 In addition to mycobacterial infections, patients with MSMD are prone to extraintestinal disease caused by nontyphoidal salmonella.
Neutrophilia
Neutrophilia is a characteristic of LAD. Other conditions associated with neutrophilia are listed in Table 51-4. Leukemoid reaction, defined as an ANC greater than 10 standard deviations above the mean for gestational age or more than 50 × 103/mm3 during the first week of life, occurs in 1.3% to 15% of infants admitted to neonatal intensive care units.120,121 It is characterized by significantly elevated numbers of early neutrophil precursors, such as myelocyte, metamyelocyte, and band forms in the peripheral blood; occasionally, promyelocytes and myeloblasts may be present. Normal proliferation, maturation, and morphology of all myeloid elements distinguish leukemoid reaction from leukemia. Toxic granules, Döhle bodies, and cytoplasmic vacuoles may be observed in the peripheral blood film in leukemoid reactions secondary to sepsis.64 Conditions associated with leukemoid reactions include prematurity,121,122 infections,123,124 severe anemia, chromosomal anomalies,125,126 exposure to antenatal corticosteroids,127,128 persistent pulmonary hypertension of the newborn,129 and bronchopulmonary dysplasia.130–132
Table 51-4 Conditions Associated with Neutrophilia in the Newborn Infant
Perinatal complications
– Infection
– Stressful labor
– Birth asphyxia
– Meconium aspiration
– Pneumothorax
– Periventricular hemorrhage
– Seizure
– Symptomatic hypoglycemia
Hematological disorders
– Hemolysis (eg, ABO incompatibility)
Leukemoid reaction
– Persistent pulmonary hypertension of the newborn (PPHN)
– Bronchopulmonary dysplasia
Chromosomal anomalies (eg, trisomy 21, trisomy 18, trisomy 13)
Leukocyte adhesion deficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


