Peroxisomal Disorders
David Valle, Gerald V. Raymond, and Stephen J. Gould
 PEROXISOME BIOLOGY AND METABOLISM
PEROXISOME BIOLOGY AND METABOLISM
Peroxisomes are single membrane-bound or-ganelles present in all cells except for erythro-cytes. In human cells, peroxisomes are spherical in shape and range in number from a few hundred to a few thousand per cell.1,2 They contain a dense proteinaceous matrix composed of 50 or more enzymes that participate in a variety of metabolic processes.3 Prominent among these is a set of enzymes catalyzing β-oxidation that are analogous to but distinct from those catalyzing mitochondrial β-oxidation and are encoded by different genes. The β-oxidation systems of peroxisomes and mitochondria have distinct but overlapping substrate specificities, with the peroxisomal system oxidizing very-long-chain (C20-C26) and long-chain (C12–C18) fatty acids and the mitochondrial system oxidizing long-chain (C12–C18), medium–chain (C12–C6), and short-chain (C6–C4) fatty acids. An additional difference is that the FAD-linked acyl-CoA oxidase, which catalyzes the first step in the peroxisomal β-oxidation spiral, is reoxidized by molecular oxygen to produce H2O2 while the analogous enzymes in mitochondria transfer their electrons to the respiratory chain via electron transport flavoprotein (ETF) and ETF-dehydrogenase. The H2O2 produced in the first step of the peroxisome β-oxidation spiral and by other peroxisomal oxidases is efficiently eliminated by catalase, another peroxisomal matrix enzyme. Additional metabolic processes that involve peroxisomal matrix enzymes include glyoxy-late transamination; β-oxidation of phytanic acid and other β-methyl-branched fatty acids; and synthesis of cholesterol, bile acids, and ether lipids such as plasmalogen and lysine degradation.
 GENETIC DISORDERS OF PEROXISOMES: OVERVIEW
GENETIC DISORDERS OF PEROXISOMES: OVERVIEW
Peroxisomal disorders can be divided into two classes: (1) peroxisomal biogenesis disorders (PBD), which are characterized by deficiency of multiple peroxisomal functions, and (2) single-function disorders in which only one peroxisomal function is deficient.1 The PBD are a genetically heterogeneous set of disorders comprising at least 13 complementation groups as determined by somatic cell hybridization studies.1,2,4 All are inherited as autosomal recessive traits and have an aggregate frequency of about 1 in 50,000. Zellweger syndrome (ZS) and rhizomelic chondrodysplasia punctata (RCDP) are examples of the PBD clinical phenotypes. At the cellular level, most PBD complementation groups demonstrate aberrant cytosolic localization of matrix proteins with relatively empty peroxisomes and normal import of peroxisomal membrane proteins and synthesis of peroxisomal membranes. By contrast, a few PBD complementation groups have no detectable peroxisomes or peroxisomal membrane vesicles.
The single-function peroxisome disorders include at least 11 different disorders inherited either as autosomal or X-linked recessive traits, nearly all of which are uncommon, with frequencies of less than 1 in 50,000.3,5 The clinical example for this class of peroxisomal disorders is X-linked adrenoleukodystrophy (X-ALD), a neurological disorder with abnormal accumulation of very-long-chain fatty acids (VLCFA) and with an incidence in males of about 1 in 20,000.6,7 At the cellular level, peroxisomes in the single-function disorders appear normal and have normal import of matrix proteins.
PEROXISOME BIOGENESIS DISORDERS (PBD)
 CLINICAL PHENOTYPES
CLINICAL PHENOTYPES
The clinical consequences of the PBD can be organized into two broad phenotypic spectra. The largest of these, the Zellweger spectrum, accounts for about 80% of PBD patients and includes at least three phenotypes originally thought to represent discrete disorders but which are now recognized as segments of a continuous spectrum.1,4,8 Listed from the most to the least severe, these three disorders are Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), and infantile Refsum disease (IRD). The second PBD phenotypic spectrum, accounting for about 20% of PBD patients, is rhizomelic chondrodysplasia punctata (RCDP). The phenotype of most RCDP patients is severe and relatively uniform, but milder variants have been described.
ZELLWEGER SPECTRUM
Zellweger syndrome, a metabolic disorder with dysmorphic features (see Table 176-1), represents the severe end of the Zellweger spectrum. These infants have a characteristic facial appearance, with a high forehead, epicanthal folds, a small nose with a broad nasal bridge, anteverted nares, and micrognathia (Fig. 162-1A and B). The anterior fontanelle is large. Cataracts and a pigmentary retinopathy are common. There is profound hypotonia, feeding problems, and growth failure. Most have neonatal seizures. Liver function is abnormal, with conjugated hyperbilirubinemia. Radiological examination reveals punctate calcifications (“calcific stippling”) in the patella and epiphyses of the long bones. Multiple small renal cysts are common but may not be detected by ultrasound examination. Abnormalities in neuronal migration (neocortical dysplasia and cerebral/cerebellar white matter disease) are common. Infants with ZS rarely live to be 1 year of age.
Neonatal adrenoleukodystrophy (NALD) is similar to, but less severe than, ZS. Dysmorphic facial features are less severe or may even be absent (Fig. 162-1C). Hypotonia and seizures are common. Because of their flat facial features and hypotonia, NALD patients are sometimes thought to have Down syndrome. Survival ranges from several months to several years. The older patients have profound mental retardation often accompanied by sensorineural hearing loss and retinopathy.
Infantile Refsum disease (IRD) patients have mild dysmorphic features and hypotonia (Fig. 162-1D). The predominant manifestations may be hepatomegaly, cholestasis, osteoporosis, and failure to thrive in early infancy. As they get older, virtually all IRD patients develop sensorineural hearing loss and pigmentary retinopathy. They usually learn to walk but have severe mental retardation. Patients with IRD may live into adulthood.
Milder variants of the Zellweger spectrum with normal development and appearance may present in adult life with sensorineural hearing loss and pigmentary retinopathy.
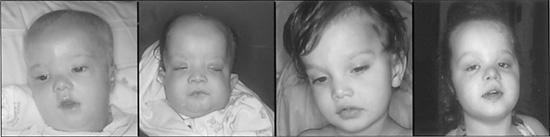
FIGURE 162-1. Facial appearances of PBD patients in the Zellweger spectrum. (A) Four-month old infant with Zellweger syndrome; (B) 3-month-old infant with Zellweger syndrome; (C) 2-year-old child with neonatal adrenoleukodystrophy (NALD); (D) 4-year-old child with infantile Refsum disease (IRD).
RHIZOMELIC CHONDRODYSPLASIA PUNCTATA SPECTRUM (RCPD)
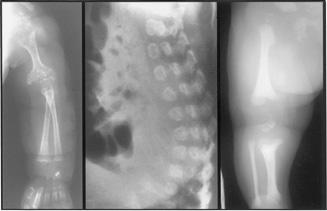
Patients with classic RCDP (see Table 176-1) have severe skeletal involvement at birth that distinguishes them from those in the Zellweger spectrum (Fig. 162-2). There is rhizomelia (shortening of the proximal limbs) and limited range of movement of the large joints of the extremities. Radiological examination shows extensive calcific stippling that involves the epiphyses of long bones; it is most prominent in the knees, elbows, hips, and shoulders. Coronal clefts of the vertebral bodies are apparent on lateral spine films. RCDP patients also have a flat face with frontal bossing. Cataracts are common, and an ichthyotic skin rash may develop after birth. Severe psy-chomotor retardation is present, and most die before 2 years of age. In addition to this classic RCDP phenotype, mildly affected patients with little or no rhizomelia have been described; some have mild intellectual defects as their only manifestation. Classic RCDP is a PBD caused by mutations in PEX7, the gene encoding the receptor for PTS2 proteins. A few patients with the classic RCDP phenotype (< 10% of the total number of patients) have single-function defect in PTS2-targeted peroxisomal matrix enzymes necessary for normal synthesis of plasmalogen.
 BASIC DEFECTS
BASIC DEFECTS
Over the last two decades, research in model organisms and PBD patients has identified a set of genes and their protein products, termed PEX genes and peroxins, respectively, that are necessary for peroxisome assembly.1,4 Most of these are involved in the targeting and uptake of matrix proteins into the organelle. All of the PBD complementation groups so far defined are explained by mutations in these PEX genes, one particular gene for each complementation group. The number of PEX genes is greater than the number of complementation groups, suggesting there are new groups to recognize. See Weller et al for a review of this topic.4
Peroxisome matrix proteins are synthesized on free cytosolic ribosomes and directed to the organelle by targeting sequences of two types (Fig. 162-3). Peroxisome-targeting signal 1 (PTS1), a C-terminal tripeptide (-SKL or conservative variants thereof), is utilized by more than 90% of the matrix proteins. PTS2, located 5 to 10 residues from the N-terminus, is utilized by a few peroxi-somal matrix proteins, including one involved in β-oxidation (peroxisomal thiolase 1), one involved in α-oxidation, and one involved in plasmalogen synthesis. The PTS1 or PTS2 motifs are bound by specific cytosolic receptors encoded by PEX5 and PEX7, respectively. Docking of the receptor and its bound cargo on the peroxisome membrane is mediated by binding to specific peroxisomal membrane proteins (PEX13, PEX14, PEX17). With the action of additional peroxisomal membrane proteins (PEX2, PEX8, PEX10, PEX12), the newly synthesized matrix proteins are translocated into the organelle, and the receptors are recycled to the cytosol. Recycling of PEX5 requires an unusual monoubiquitination on a conserved cysteine near the N-terminus of the protein.9 The recycling process is facilitated by the action of PEX1, PEX6, PEX22, PEX4, and PEX26. Genetic defects in this subset of PEX genes are responsible for nine PBD complementation groups that are characterized at the cellular level by mislocalization of matrix proteins to the cytosol.
The mechanism and signals involved in targeting peroxisomal membrane proteins to the organelle are less well understood, although PEX genes encoding proteins that function in this process have been identified (PEX3, PEX16, PEX19).4 Not surprisingly, genetic defects in this subset of peroxins are responsible for the three PBD complementation groups characterized by lack of detectable peroxisomes.
Recently, an infant with metabolic features consistent with abnormal peroxisome and mi-tochondrial function has been described and represents a different type of PBD.10 In this patient’s cells, there was a defect of organellar fission of both peroxisomes and mitochondria caused by a dominant negative mutation in the dynamin-like protein 1 gene DLP1. This observation, together with studies in model systems, indicates that normal DLP1 function is required for the division of two different subcellular organelles.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


