Fig. 26.1
Brain tissue O2 tension, as well as cerebral perfusion, is progressively increased with increases in inspired CO2 concentration (Reproduced with permission, Hare et al. (2003))
26.2.3 Cardiovascular System
Hypercapnic acidosis has complex opposing effects on the cardiovascular system. Activation of the sympathoadrenal axis results in indirect effects including increases in preload, heart rate, and myocardial contractility, as well as reduced afterload. These indirect effects are in part offset by direct reduction of cardiac and vascular smooth muscle contractility (Tang et al. 1991), with a net increase in cardiac output (Cullen and Eger 1974).
The complex interaction of hypoxic pulmonary vasoconstriction, intrapulmonary shunt, and elevated cardiac output ultimately results in increased global and regional O2 delivery (Cardenas et al. 1996). Tissue oxygen delivery is enhanced by hypercapnic acidosis (above) increasing subcutaneous and intestinal O2 tension (Ratnaraj et al. 2004) and presumably in multiple other systemic tissues also.
26.3 Insights from Laboratory Studies
The effects of hypercapnia and acidosis in relevant preclinical models of lung and systemic organ injury are increasingly well understood. A clear understanding of these effects – both beneficial and deleterious – should guide the rational use of hypercapnia in the clinical setting. In addition, such knowledge will provide insights into the potential contribution of hypercapnia to the beneficial effects of low tidal volume ventilation.
26.3.1 Pulmonary
Hypercapnic acidosis has been demonstrated to have independent beneficial effects in the setting of acute lung injury following ischemia-reperfusion, free radical injury, mechanical stretch, and hypoxia-induced pulmonary hypertension. Hypercapnic acidosis, generated by increasing the inspired CO2, directly attenuates acute lung injury induced by ischemia-reperfusion (Shibata et al. 1998; Laffey et al. 2000a) and free radical mechanisms (Shibata et al. 1998). In contrast, hypocapnic alkalosis worsens reperfusion-induced lung injury (Laffey et al. 2000b; Laffey and Kavanagh 2002). In in vivo pulmonary ischemia-reperfusion, hypercapnic acidosis lessened protein leak and preserved lung mechanics. These effects then resulted in subsequent improvement in oxygenation with hypercapnic acidosis as compared to normocapnia (Laffey et al. 2000c). Hypercapnic acidosis has also been demonstrated to attenuate lung injury following mesenteric ischemia-reperfusion (Laffey et al. 2003a), which occurred despite the associated elevations in pulmonary arterial pressures.
Hypercapnic acidosis has benefit in chronic hypoxia-induced pulmonary hypertension in newborn rats (Kantores et al. 2006). In this model, animals were randomized (at birth) to 0, 5.5, or 10 % added CO2 and were also randomized to either normoxia (21 % O2) or moderate hypoxia (13 % O2). Pulmonary hypertension (characterized by increased pulmonary arterial resistance, right ventricular hypertrophy, right ventricular dysfunction, pulmonary artery smooth muscle hypertrophy) was reliably induced by chronic hypoxia but was attenuated by inspired CO2. Indeed, a dose response was seen between level of inspired CO2 and the inhibition of pulmonary vascular remodeling, while both 5 and 10 % inspired CO2 normalized right ventricular dysfunction. Finally, exposure to 10 % CO2 also reduced oxidative lung injury and prevented upregulation of endothelin-1, a critical mediator of pulmonary vascular remodeling (Kantores et al. 2006).
The effects of hypercapnic acidosis in stretch-induced lung injury are more complex. Two key studies have demonstrated that the addition of inspired CO2 attenuated Ventilator-induced Lung Injury (VILI) in the ex vivo (Broccard et al. 2001) and in vivo (Sinclair et al. 2002) rabbit lung. However, the benefits of supplemental CO2 are not as pronounced in models where tidal volumes used are comparable to clinical values (i.e., in terms of mL/kg of body weight). For example, one model used a clinically relevant high tidal volume strategy (12 mL/kg) and demonstrated that in this context, hypocapnia was potentially deleterious and hypercapnic acidosis somewhat protective (Laffey et al. 2003b). In contrast, in an atelectasis-prone model of lung injury, which may in part reflect neonatal respiratory distress syndrome, supplemental CO2 did not lessen the injury (Rai et al. 2004). Furthermore, when hypercapnia was induced by reducing the respiratory rate (i.e., not by reducing the tidal volume or by adding CO2 to the inspired gas), there was evidence of harm (Billert et al. 2003). Thus the benefits of hypercapnia appear to depend on the severity of lung stretch, with hypercapnia more protective in the setting of more severe stretch. The means of producing the hypercapnia also appears to be important, with inspired CO2 being protective, while hypercapnia produced simply by reducing respiratory rate is potentially deleterious.
In addition, hypercapnia may impair the healing process following lung injury. In this regard, Doerr et al. reported that, while hypercapnia minimized the adverse effects of high-volume ventilation on vascular barrier function, it also impaired the ability of lung cells to repair following high-stretch injury (Doerr et al. 2005). Hypercapnia – both in the presence (O’Toole et al. 2009) and absence of acidosis (O’Toole et al. 2009; Nichol et al. 2009) – inhibits healing of pulmonary epithelial wounds in vitro. The mechanisms underlying this inhibitory effect of hypercapnia on wound healing appear to involve inhibition of the activity of nuclear factor kappa B (NF-κB), a pivotal transcriptional activator in the setting of injury and repair (O’Toole et al. 2009).
Hypercapnia may also have a negative impact on diaphragmatic function, as it interferes with neuromuscular transmission and causes a reduction in cross-sectional area of type IIb fibers (Shiota et al. 2004). Whether this interference with neuromuscular transmission translates into an important clinical effect remains to be proven. In fact permissive hypercapnia in one study was shown to augment weaning from mechanical ventilation (Mariani et al. 1999).
The potential for hypercapnia to impair the immune system response to live bacterial sepsis (Laffey et al. 2004a; Swenson 2004) is of concern given that lung injury or ARDS commonly develops in the context of severe sepsis (Dahlem et al. 2003). Consequently, determination of the effects of hypercapnia and acidosis in this setting has been the focus of much recent work. When endotoxin – the toxin produced by gram-negative bacteria – is administered by the intratracheal route, hypercapnic acidosis delivered via inspired CO2 is protective (Laffey et al. 2004b). In contrast, hypercapnia produced by reduced respiratory rate appears to worsen the severity of lung injury produced by systemic endotoxemia (Lang et al. 2005). Of perhaps greater relevance to the clinical setting, more recent work has demonstrated beneficial effects in animal models of severe acute pneumonia (Ni Chonghaile et al. 2008a) as well as established severe pneumonia (Ni Chonghaile et al. 2008b). However, hypercapnic acidosis did not modulate the severity of less severe pneumonia-induced lung injury (O’Croinin et al. 2005). Of more concern, sustained environmental hypercapnia worsened the severity of lung injury induced by prolonged bacterial pneumonia (O’Croinin et al. 2008). To summarize, the effects of hypercapnic acidosis in models of pneumonia depend on the severity of pneumonia and the methods by which hypercapnia is attained.
The effects of hypercapnia and acidosis in the setting of systemic sepsis are increasingly well understood. Hypercapnic acidosis attenuated shock and acute lung injury cecal ligation and puncture (Costello et al. 2009). These protective effects were seen in both the early phases and in the setting of prolonged systemic sepsis (Costello et al. 2009). Interestingly, buffering of the hypercapnic acidosis attenuated its protective effects on the lung, while preserving its beneficial hemodynamic effects (Higgins et al. 2009). These studies provide some reassurance regarding the safety of permissive hypercapnia in the setting of sepsis but there is also evidence of the potential for hypercapnic acidosis to have a negative impact in sepsis. In summary, the effects of hypercapnia and acidosis appear to depend on the severity and duration of the septic insult, and also to some extent on the primary site of sepsis.
26.3.2 Cardiovascular
The benefits of hypercapnic acidosis in myocardial ischemia-reperfusion are well characterized (Nomura et al. 1994). However, the associated protection may be a function of the acidosis per se (as opposed to the elevated PCO2) since reduction in in vivo myocardial infarct size occurs whether acidosis results from elevated CO2 or from metabolic causes (Kitakaze et al. 1988, 1997; Preckel et al. 1998). However, increased coronary blood flow is also seen with hypercapnic acidosis (Nomura et al. 1994; Arellano et al. 1999), and this may confer additional benefits beyond those observed with metabolic acidosis. However, on the negative side, the presence of hypercapnic acidosis may reduce the likelihood of successful resuscitation following experimental ventricular fibrillation (von Planta et al. 1991).
26.3.3 Neurologic
While an increasing FiCO2 can improve cerebral oxygenation in a dose-dependent manner in anesthetized rats (Hare et al. 2003), the effect of CO2 in hypoxic-ischemic brain injury models is dose dependent. Hypercapnic acidosis does protect against hypoxic-ischemic injury in the immature brain in porcine (Barth et al. 1998; Rehncrona et al. 1989) and rodent (Vannucci et al. 1995, 1997) models. However, the most benefit occurred in the mild to moderate range of hypercapnia (PaCO2 40–55 mmHg), as compared to higher levels of PaCO2 (e.g., >70 mmHg) (Vannucci et al. 1995). Indeed, even higher levels of PaCO2 that are not generally seen in the clinical setting may be detrimental to the immature brain (Vannucci et al. 2001). Reasons for protective effects of hypercapnia in hypoxic brain injury include free radical inhibition (Barth et al. 1998; Rehncrona et al. 1989), attenuation of neuronal apoptosis, and reduction in the CNS levels of glutamate (a neurotransmitter that can cause excitotoxicity) (Vannucci et al. 1997).
Hypercapnia has the potential to contribute to retinopathy of prematurity since it has been shown experimentally to produce preretinal neovascularization similar to that seen in oxygen-induced retinopathy in a neonatal rat model (Holmes et al. 1998). This is of obvious concern in the clinical setting of neonatal respiratory failure.
26.3.4 Others
The response of cells to hypercapnia and hypoxia unsurprisingly depends on their origin. For instance, renal tubular cells undergo apoptosis in response to hypoxia/hypercapnia (Hotter et al. 2004). In contrast, isolated hepatocytes exposed to anoxia (Bonventre and Cheung 1985) and chemical hypoxia (Gores et al. 1989) show improved survival under acidotic conditions which may be an adaptive phenomenon against hypoxic-ischemic stress.
26.4 Cellular and Molecular Effects of Hypercapnia
To anticipate potential pitfalls of permissive hypercapnia and to complete translational research from bench to bedside, it will eventually be important to have a basic understanding of the cellular and biochemical effects of hypercapnia. In keeping with the trend towards individualized medicine, increasing appreciation of the underlying molecular mechanisms may allow the clinical practitioner to ultimately identify those situations in which permissive hypercapnia may be beneficial. Knowledge of molecular mechanisms of hypercapnia will also help identify the circumstances in which it may cause harm.
26.4.1 Acidosis vs. Hypercapnia
The protective effects of hypercapnic acidosis appear to be a function of the acidosis generated as opposed to hypercapnia per se (Laffey et al. 2000a; Higgins et al. 2009; Lang et al. 2000). The beneficial effects of hypercapnic acidosis in ischemia-reperfusion in an isolated lung model were ablated if the pH was buffered towards normal (Laffey et al. 2000a). Furthermore, buffering of the hypercapnic acidosis abolished the protective effects on the lung in the setting of systemic sepsis (Higgins et al. 2009). Metabolic acidosis demonstrates protective effects in many models of organ injury including the myocardium (Kitakaze et al. 1988, 1997; Preckel et al. 1998), liver (Bonventre and Cheung 1985), and kidney (Bonventre and Cheung 1985). In the latter, isolated renal cortical tubules exposed to anoxia have better conserved ATP status where reoxygenation occurs in an acidotic (vs. alkalotic) environment (Bonventre and Cheung 1985). However, the inhibition of wound healing by hypercapnic acidosis in experimental models be it cell culture (O’Toole et al. 2009) or in an ex vivo model (Caples et al. 2009) would appear to be independent of buffering.
26.4.2 Regulation of Gene Expression
Hypercapnic acidosis affects a key transcriptional factor, nuclear factor kappa B (NF-κB), which is an important regulator of the expression of multiple genes that play a central role in inflammation and repair. Hypercapnic acidosis inhibits endotoxin-induced NF-κB activation and DNA binding in pulmonary endothelial cells by decreasing IκB-α degradation (Fig. 26.2) (Takeshita et al. 2003). Endothelial production of intercellular adhesion molecule ICAM-1 and interleukin-8 are downstream effects, reflecting suppression of NF-κB activation by hypercapnic acidosis. While inhibition of NF-κB can have anti-inflammatory effects, inhibition may also effect wound healing. Hypercapnic acidosis was shown to inhibit pulmonary wound healing via an NF-κB-dependent mechanism (O’Toole et al. 2009).
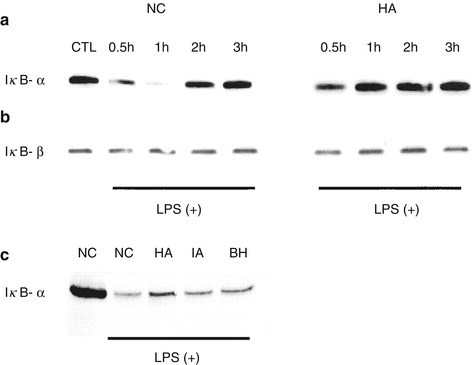
Fig. 26.2
Hypercapnia suppresses the degradation of IκB-α (Panel a) but not IκB-β (Panel b) following exposure to lipopolysaccharide, thereby inhibiting the nuclear translocation of NFκ-B and downstream cytokine production. The effects of isocapnic acidosis and buffered hypercapnia (Panel c) on IκB-α degradation were intermediate between normocapnic control and hypercapnic acidosis conditions. Symbols: BH buffered hypercapnia, HA hypercapnic acidosis, IA isocapnic acidosis, LPS lipopolysaccharide, NC normocapnia, NFκ-B nuclear factor kappa B (Reproduced with permission, Takeshita et al. (2003))
26.4.3 Attenuation of Neutrophil Activity and Cytokine Levels
Another key component of the inflammatory response which is inhibited by hypercapnic acidosis is neutrophil function. Hypercapnic acidosis interferes with neutrophil intracellular pH regulation (Hackam et al. 1996), and this is associated with attenuation of important neutrophil functions such as chemotaxis (Demaurex et al. 1996) and release of interleukin-8 (IL-8) following LPS stimulation (Coakley et al. 2002). These effects are also seen in vivo where lung neutrophil recruitment is inhibited during ventilator-induced (Sinclair et al. 2002) and endotoxin-induced (Laffey et al. 2004b) lung injury. Hypercapnic acidosis has been demonstrated to reduce neutrophil phagocytic activity (O’Croinin et al. 2008). Finally, hypercapnic acidosis inhibits other proximal inflammatory responses, such as production of TNF-α following in vivo pulmonary ischemia-reperfusion (Laffey et al. 2000c) as well as release of TNF-α and IL-1, release from stimulated in vitro macrophages (West et al. 1996).
26.4.4 Modulation of Free Radical Biology
The potential for hypercapnic acidosis to promote nitration of lung tissue in vivo appears to depend on the particular injury process in question. One potential problem with hypercapnic acidosis is its potential to augment tissue nitration by peroxynitrite, a potent free radical as demonstrated when buffered hypercapnia promotes the formation of nitration products from peroxynitrite in vitro (Lang et al. 2000; Zhu et al. 2004). Hypercapnic acidosis increases nitration following endotoxin exposure (Laffey et al. 2004b; Lang et al. 2000, 2005) but decreases it following pulmonary ischemia-reperfusion (Laffey et al. 2000c).
In contrast, hypercapnic acidosis appears to reduce oxidant production and oxidant-mediated tissue injury. Enzymes which produce oxidizing free radicals function optimally at physiological pH. Neutrophil activity is regulated in part by ambient CO2 levels with hypercapnia inhibiting – and hypocapnia increasing – the production of oxygen-derived free radicals (Coakley et al. 2002). Superoxide synthesis by stimulated neutrophils in vitro is decreased at acidic pH (Leblebicioglu et al. 1996). In lung ischemia-reperfusion models, hypercapnic acidosis reduces free radical tissue injury (Laffey et al. 2000c) and inhibits the production of higher oxides of NO following both ventilator-induced (Broccard et al. 2001) and endotoxin-induced (Laffey et al. 2004b) lung injury. In the brain, glutathione depletion and lipid peroxidation – reflecting free radical activity– and resultant tissue damage are attenuated by hypercapnic acidosis (Barth et al. 1998).
26.5 Role of Permissive Hypercapnia in Clinical Settings
Once ventilator-induced lung injury has occurred, there are currently no available therapies to reverse it; thus, the focus in recent years has been on minimizing ventilator-induced lung injury. Since the recognition that end-inspiratory distension may be a prime determinant of lung injury (Dreyfuss and Saumon 1998), avoidance of high tidal volumes has become a focus of management. While increasing respiratory rate can control the PaCO2, the mechanical properties of the injured lung are a limiting factor since they determine the time necessary for inspiration and expiration. Thus permissive hypercapnia will occur using low tidal volumes, particularly as lung compliance is lowered. But is permissive hypercapnia beneficial – or even safe – in specific pediatric clinical scenarios?
26.5.1 Neonatal Respiratory Distress Syndrome
Mariani et al. reported beneficial effects of permissive hypercapnia in 49 infants with neonatal respiratory distress syndrome in the first randomized controlled trial of permissive hypercapnia (Mariani et al. 1999). Preterm infants randomized to permissive hypercapnia received lower tidal volumes, and they required fewer days of assisted ventilation compared with children (mean 2.5 vs. 9.5 days) (Fig. 26.3). No obvious adverse effects were seen, but the study was not powered adequately to draw conclusions regarding safety. However, the use of postnatal steroids, total duration of oxygen supplementation, and incidence of air leaks, reintubations, and chronic lung disease did not differ between those randomized to permissive hypercapnia or conventional ventilation (Fig. 26.3).
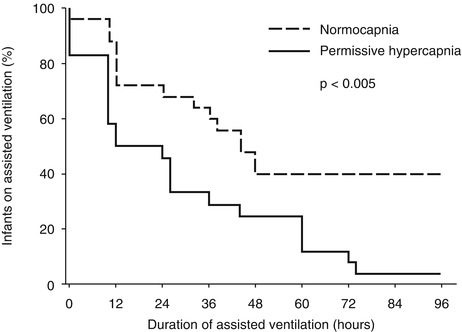
Fig. 26.3
Duration of mechanical ventilation in neonates with respiratory failure randomized to conventional therapy or permissive hypercapnia (Reproduced with permission, Mariani et al. (1999))
A multicenter trial by the National Institutes of Child Health and Development Neonatal Network randomized extremely low-birth-weight infants ventilated before 12 h of life to normocapnia or permissive hypercapnia as well – in a factorial design – to dexamethasone vs. placebo (Carlo et al. 2002). The study, however, was stopped early after enrolment of 220 patients because of an unexpected increased incidence of bowel perforation in infants receiving early dexamethasone. Nonetheless, there was a trend towards a lower incidence of death and chronic lung disease with permissive hypercapnia, and only 1 % of permissive hypercapnia group patients required mechanical ventilation at 36 weeks, compared to 16 % in the control group. No adverse effects were specifically associated with permissive hypercapnia; in fact, infants receiving permissive hypercapnia appeared to have a lower incidence of severe intraventricular hemorrhages (20 % vs. 26 %) (Carlo et al. 2002).
The most recent trial in 2006 was again stopped prematurely after enrolment of only 40 patients when the permissive hypercapnia group showed nonsignificant trends towards increased mortality and poorer neurological outcome (Thome et al. 2006). Thus what little evidence is available in this particular setting is conflicting, but the uncertainty highlights the need for further understanding of the mechanisms and effects of hypercapnia before additional large randomized controlled clinical trials are undertaken.
26.5.2 Persistent Pulmonary Hypertension of the Newborn (PPHN)
The poor neurological outcome associated with PPHN can be explained in part by a traditional approach of using hyperventilation (and targeted hypocapnia) to lower pulmonary arterial pressure (Drummond et al. 1981; Ferrara et al. 1984; Leavitt et al. 1987). In one small series of 15 infants where treatment focused on minimizing barotrauma (allowing PaCO2 as high as 60 mmHg), all children survived, and only a single patient developed chronic lung disease (Wung et al. 1985). In another series of 34 infants with severe PPHN managed with permissive hypercapnia, Marron et al. reported 100 % survival, with good neurologic outcome (Marron et al. 1992).
26.5.3 Congenital Diaphragmatic Hernia
Ventilation strategies which involve achieving systemic alkalinization through hyperventilation may result in poor long-term outcomes. Thus permissive hypercapnia has begun to play a more prominent role in the management of congenital diaphragmatic hernia (Bohn 2002). In a recent retrospective analysis, Bagolan et al. reported that strategies allowing permissive hypercapnia to develop were beneficial (Bagolan et al. 2004). Increased survival and reduced barotrauma and morbidity were associated with the use of permissive hypercapnia. However, the use of high-frequency oscillation provides a mechanism for controlling (i.e., lowering) PaCO2 without the use of high tidal volumes. In this setting, it is not clear whether hypocapnia or high tidal volume (or both) are responsible for adverse pulmonary (or CNS) outcome.
26.5.4 Congenital Heart Disease
Low cerebral blood flow has been associated with periventricular leukomalacia and adverse neurologic outcome in neonates with severe congenital heart defects (Licht et al. 2004). Thus preventing reduced cerebral perfusion – or augmenting it – would be desirable. Licht et al. showed significant deficits in cerebral blood flow that were reversed when CO2 was administered in small series of 25 infants (Licht et al. 2004). Also, the addition of inspired CO2 increased cerebral oxygenation and mean arterial pressure compared with reducing FiO2 in hypoplastic left heart syndrome (Tabbutt et al. 2001) and following cavopulmonary connection (Ramamoorthy et al. 2002), respectively.
Following cavopulmonary connection, traditional management has focused on avoiding hypercapnia because of the fear that hypercapnia would increase pulmonary vascular resistance and reduce systemic oxygenation. Recent evidence has demonstrated that hypoventilation-induced hypercapnia actually increases pulmonary perfusion primarily through augmentation of systemic blood flow and a reduction of systemic vascular index (Bradley et al. 2003). In addition, allowing PaCO2 to return to 40 mmHg following hypercapnia decreases pulmonary blood flow and systemic oxygenation following cavopulmonary connection (Hoskote et al. 2004).
26.5.5 Acute Severe Asthma
Darioli and Perret were the first to describe avoidance of lung injury through lowering tidal volume (and permitting hypercapnia) (Darioli and Perret 1984); in their series, they described mechanically ventilated patients with status asthmaticus, in whom there were no deaths in 26 patients. In status asthmaticus, plateau pressure and tidal volume are reduced and expiratory time increased in order to lower end-inspiratory lung volume and (potentially lethal) barotrauma. Such an approach can result in significant levels of hypercapnia which is usually well tolerated, although life-threatening acidosis has been managed successfully with extracorporeal arteriovenous carbon dioxide removal (Conrad et al. 2007).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


