Peripheral Nerve Disorders and Anterior Horn Cell Diseases
Peter B. Kang
The classification of peripheral nerve disorders can be tricky, as with many major disease categories. Neuropathies may be classified by etiology (genetic versus acquired), physiology (demyelinating versus axonal), anatomy (focal versus generalized, sensory versus motor), and chronicity (acute versus chronic). This chapter focuses on the major categories of peripheral nerve disorders, including anterior horn cell diseases, using a combination of the above variables to classify them.
Focal neuropathies, including brachial plexopathies, are relatively simple to localize on physical examination, especially when accompanied by a history of trauma or other clear cause. Generalized neuropathies, including anterior horn cell disorders, may be more difficult to localize neuroanatomically. A distal pattern of weakness is usually, but not always, consistent with a polyneuropathy. Distal muscle atrophy may be present. Anterior horn cell diseases are typically associated with proximal or generalized weakness. Deep tendon reflexes are diminished or absent in polyneuropathies and anterior horn cell disorders.
The term anterior horn cell disease is synonymous with motor neuron disease, as the cell body of the motor neuron lies within the anterior horn of the spinal cord. In children, the dominant motor neuron diseases are spinal muscular atrophy (SMA) and poliomyelitis (in countries where vaccination is inconsistent), whereas in adults amyotrophic lateral sclerosis (Lou Gehrig disease) is most common. The latter will not be discussed, as the juvenile form of amyotrophic lateral sclerosis is extremely rare in childhood.
SPINAL MUSCULAR ATROPHY
SMA is an inherited disorder of the motor neuron in the spinal cord, which leads to progressive weakness of the skeletal muscles, affecting the proximal muscles in the lower extremities first, followed by spread to the distal muscles and upper extremities.1 The intercostal respiratory muscles are significantly involved, but the muscles of the heart and face (including eyes) are spared. Intelligence is normal. The carrier frequency is estimated to range from 1 in 40 to 1 in 50 in both genders and all known ethnic groups.2 Based on data from several countries, the incidence is estimated to range from 1 in 6000 to 1 in 10,000 live births.3-5
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The severity of SMA varies considerably, and most affected individuals can be classified phenotypically as having type 1, 2, or 3, with some patients having intermediate forms. The original term for SMA type 1 was Werdnig-Hoffman disease, but that term is less commonly used today. The original term for SMA type 3 was Kugelberg-Welander disease, but that term has also fallen into disuse. The classification is based on motor milestones achieved: type 1 patients never sit or walk, type 2 patients sit but never walk, and type 3 patients walk for at least some period of their lives. Type 1 patients tend to have onset before 6 months, type 2 between 6 and 18 months, and type 3 after 18 months. Some clinicians recognize a severe congenital variant of type 1 as type 0 and a mild adult-onset form as type 4. Patients with types 1 and 2 usually present with a combination of hypotonia, delayed motor milestones, and feeding difficulties. Patients with type 3 may present with motor delays or gait difficulty.
Findings on physical examination are important in ascertaining the possible diagnosis of spinal muscular atrophy (SMA). Regardless of the type, deep tendon reflexes will be absent or depressed. Infants with SMA 1 and 2 will have peripheral hypotonia. Hypotonia in infants can be detected by signs such as frog leg posture, head lag, and slip-through on vertical suspension (see Chapter 569). Peripheral hypotonia is caused by a peripheral nervous system lesion and is accompanied by weakness on examination, most easily observed when the infant is upset. Central hypotonia is caused by a central nervous system lesion or generalized genetic disorder such as Down syndrome and is not accompanied by weakness. Such infants will become much more vigorous when upset. Infants with SMA 1 may also have some bulbar abnormalities, including depressed gag reflex, sucking and swallowing difficulties, and a weak cry, but otherwise have normal cranial nerve function. Children with SMA 3 also have peripheral hypotonia, but as these children present after infancy, the examination of muscle tone is similar to that in adults. The motor and reflex findings are typically symmetrical, but some exceptions have been reported.6
 DIAGNOSIS
DIAGNOSIS
The differential diagnosis varies, depending on the age, presentation, and the particular type of spinal muscular atrophy (SMA) in question. In infants and toddlers who appear to have SMA 1 or 2, congenital myopathies and congenital muscular dystrophies should be considered, although both are rarer. Many of the congenital myopathies are associated with facial weakness, ophthalmoplegia, or both. The more severe congenital muscular dystrophies are accompanied by various forms of central nervous system abnormalities. In infants with a more acute presentation of hypotonia and weakness, infant botulism may be a consideration, especially in endemic regions. Some infants with central hypotonia, such as Prader-Willi syndrome, may have hypotonia with depressed or absent reflexes, but they typically do not have weakness. The most common disorder to consider in the differential diagnosis of SMA 3 is muscular dystrophy. It is rare for a congenital myopathy to present at the age when SMA 3 is typically diagnosed. If the weakness is not clearly proximal in distribution, polyneuropathies such as Charcot-Marie-Tooth disease and polyneuropathies associated with systemic diseases should be considered.
Almost all cases of SMA are caused by a homozygous deletion in exon 7 of the SMN1 gene on chromosome 5q13, with some of the remainder caused by an exon 7 deletion in 1 allele and a point mutation in the other.7,8 Ultimately, however, SMA is defined pathologically; thus there are rare inherited cases of anterior horn cell disease in childhood that are not associated with any identifiable mutations in SMN1, often with some atypical clinical features such as arthrogryposis multiplex congenita9 The SMN1 deletion does not explain the clustering of severity into the various types. SMN2 is a similar and neighboring gene on chromosome 5q13 that differs from SMN1 by only 5 nucleotides. It is typically expressed in lower amounts than SMN1 and tends to produce a truncated version of the protein product. However, higher levels of SMN2 expression can compensate in part for the homozygous SMN1 deletion, and indeed, the number of copies of SMN2 vary from individual to individual and correlates inversely with the degree of severity in SMA.10
Before the discovery of the association of spinal muscular atrophy (SMA) with mutations in the SMN1 gene, the diagnosis during life was based on clinical presentation, neurogenic findings with sparing of the sensory nerves on electromyography (EMG) studies (Fig. 570-1), and sometimes neurogenic findings on muscle biopsy (Fig. 570-2). Creatine kinase levels are normal or mildly elevated. Postmortem confirmation could be made via histologic analysis of the spinal cord. The diagnosis of SMA during life remains a clinical one today; however, genetic testing has become widely available in a number of countries and serves as the standard for confirmation of the diagnosis. This has made muscle biopsy unnecessary in most cases. The genetic test is a simple polymerase chain reaction–based test for exon 7 deletions in the SMN1 gene. The main limitation of genetic testing is the delay in obtaining results, which may be several weeks. This delay is important, because infants affected with type 1 may have significant respiratory and feeding issues at presentation. EMG continues to be helpful in the diagnosis in centers where pediatric EMG studies are readily available. In cases of type 1, a rapid diagnosis is often beneficial for decision-making regarding interventions and resuscitation status. Cases of types 2 and 3 may mimic other diseases such as muscular dystrophy. Additionally, rare cases may present with atypical features such as asymmetric weakness and upper motor neuron signs.6
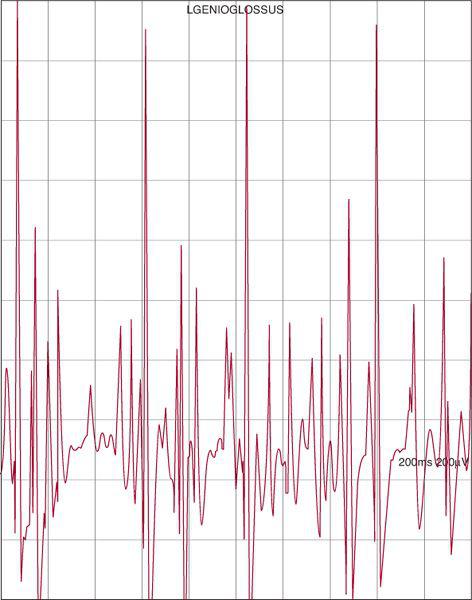
FIGURE 570-1. Left genioglossus needle electromyography (EMG) in a 3-week-old girl with peripheral hypotonia, areflexia, and episodic hypoxia demonstrates a large, rapidly firing motor unit (≥ 2 mV amplitude, approximately 20 Hz) consistent with a neurogenic injury. Subsequent genetic testing confirmed a homozygous deletion in exon 7 of SMN1. Horizontal divisions 20 ms, vertical divisions 200 μV.
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
The prognosis varies widely. In type 1, affected individuals often do not live longer than 2 years; however, with respiratory interventions such as noninvasive positive-pressure ventilation and gastrostomy tube feedings, children may live several years longer. One patient in our clinic with type 1 has survived for 20 years, but this is exceptional. Patients with type 2 typically survive to early adulthood, whereas patients with type 3 have a normal or only slightly shortened life expectancy. The major life-threatening complication in all types is respiratory failure. Nutritional issues also pose a significant problem, especially for type 1 patients and some type 2 patients.
Supportive therapies can improve the quality of life and life expectancy. Respiratory interventions include cough-assist machines, supplemental oxygen, noninvasive positive-pressure ventilation, and in rare cases, tracheostomy and chronic mechanical ventilation. The nutritional problems may be ameliorated by nasogastric or gastrostomy tube feedings for patients with types 1 and 2. Physical therapy, orthotic devices, and orthopedic surgical procedures may be helpful. Genetic counseling is important, as most parents of affected children have a 25% chance of having another affected child with each subsequent pregnancy. Interventions such as preimplantation genetic testing, which requires in vitro fertilization, may be used to minimize the likelihood of recurrence.
Researchers are investigating a variety of experimental therapies. Among these, histone deacetylase (HDAC) inhibitors such as hydroxyurea, valproate, and butyrate compounds have recently received the most attention, especially since they are already approved by the US FDA for other indications. HDAC inhibitors seem to trigger an increase in full-length SMN protein expression from SMN2 templates.11 Several trials of HDAC inhibitor therapy in spinal muscular atrophy have been conducted in humans, with some promising but preliminary results.
POLIOMYELITIS
Poliomyelitis is an RNA virus spread by the oral-fecal route. It was previously a devastating epidemic disease, but since the 1950s the widespread institution of immunization programs has essentially eradicated this disorder in most nations. The few cases of vaccine-associated paralytic polio in the United States have been eliminated in recent years because children currently receive inactivated poliovirus vaccine rather than the oral poliovirus vaccine. However, polio has proven to be difficult to eradicate on a global scale. Reservoirs of the virus persist in rare asymptomatic humans and in laboratories, and small outbreaks continue to occur in regions where vaccination is inconsistent. Some children with a history of prior infection migrate from those areas to the United States and other developed nations, either through immigration with their families or through adoption. Direct evidence for acute polio infection can be obtained by enteroviral isolation from throat and stool swabs, but a prior polio infection may be difficult to confirm.12
Paralytic poliomyelitis (also known as infantile paralysis) occurs in a small fraction of polio cases, leaving significant and permanent motor neuron damage in its wake. A hallmark of remote poliomyelitis is asymmetric weakness and muscle atrophy of 1 or more limbs, often accompanied by leg-length discrepancy. The diagnosis of prior poliomyelitis can be supported by EMG findings, which demonstrate neurogenic abnormalities, sparing the sensory nerves, as in other anterior horn cell diseases. Therapy is supportive and may include physical therapy, shoe adjustments and other orthotic devices, and orthopedic surgical interventions. Decades after the original infection, patients may develop a post-polio syndrome, in which weakness may abruptly worsen in the affected extremities, and then stabilize again.
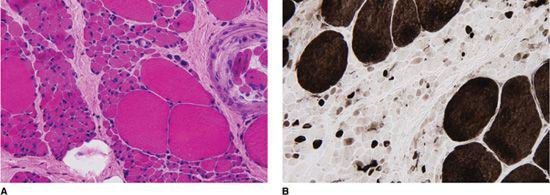
FIGURE 570-2. Vastus lateralis muscle biopsy demonstrating neurogenic features in a 4-year-old girl with gait difficulties, frequent falls, proximal weakness, and diminished deep tendon reflexes. A: Frozen section, hematoxylin & eosin stain, 40× magnification. Note sheets of small extremely atrophic fibers (12–15 μm), as well as clusters of large hypertrophic fibers (120 μm). A muscle spindle is present on the right edge of the field. B: An ATPase histochemical stain at pH 4.3, visualized at 40× magnification, demonstrates that the small atrophic fibers are both type I and type II, but the large hypertrophic fibers are type I, characteristic of spinal muscular atrophy. (Courtesy of Hart GW Lidov, MD, PhD, Department of Pathology, Children’s Hospital Boston, Boston, MA.)
INHERITED NEUROPATHIES (CHARCOT-MARIE-TOOTH DISEASE)
Charcot-Marie-Tooth (CMT) disease is a broad class of inherited peripheral neuropathies. They are also known as hereditary motor and sensory neuropathies, but the traditional term CMT remains the dominant one. The exact prevalence in large populations is difficult to estimate, as most epidemiologic studies were performed before the various genetic etiologies were discovered, but several of those studies found a prevalence of approximately 30 per 100,000.13,14
There are several major subcategories of CMT. CMT1 includes demyelinating polyneuropathies with autosomal dominant inheritance, and it is by far the most common group. CMT2 includes axonal polyneuropathies with autosomal dominant inheritance. Dejerine-Sottas disease, an early-onset form, was previously classified as CMT3, but is now more commonly known by the eponym. CMT4 includes the autosomal recessive forms, with either demyelinating or axonal physiologies. CMT disease X is the X-linked form, which has both demyelinating and axonal physiology.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
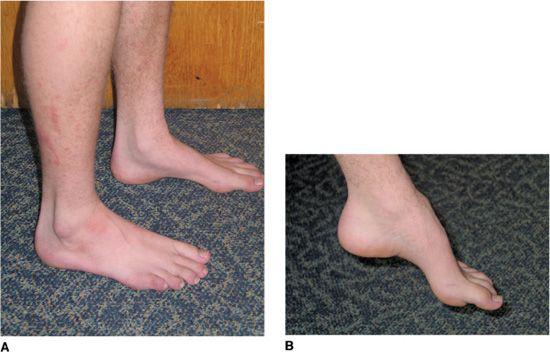
The age of onset varies by subtype, but many affected individuals present in childhood or adolescence with slowly progressive gait difficulties, sometimes accompanied by frequent falls, although some may not come to attention until adulthood. Physical examination is notable for pes cavus, hyporeflexia or areflexia, and symmetrical distal weakness with foot-drop and atrophy (Fig. 570-3). The footdrop and atrophy may not be evident early in the course. Sensory loss is often present on examination, although the patients may not complain of this. A tremor may be present. Some individuals present for evaluation due to a positive family history, even when they have no symptoms.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


