Other Disorders of Amino Acid Metabolism
Jean-Marie Saudubray
 IMIDAZOLE-DIPEPTIDE METABOLISM DEFECTS: CARNOSINE AND HOMOCARNOSINEMIA
IMIDAZOLE-DIPEPTIDE METABOLISM DEFECTS: CARNOSINE AND HOMOCARNOSINEMIA
The enzyme defects, clinical manifestations, metabolic disturbances, and treatment of these disorders are presented in Table 144-1.
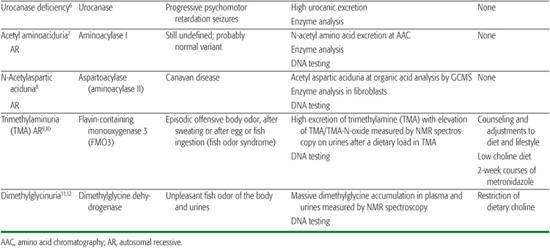
Table 144-1. Miscellaneous Amino Acid Disorders
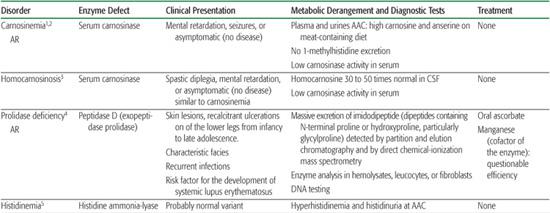
Imidazole dipeptides derive their name from the imidazole ring of histidine. Carnosine (beta alanine histidine) is found in skeletal muscles and the brain, where it may be a neurotransmitter. It is hydrolyzed by two isozymes. Cytosolic carnosinase displays a very broad dipeptidase activity but does not hydrolyze anserine or homocarnosine. Serum carnosinase, also found in the cerebrospinal fluid, hydrolyzes carnosine and anserine but hydrolyzes homocarnosine very poorly. Anserine (beta alanine-1-methylhistidine) is normally absent from human tissues and body fluids but may be derived from the diet and is found in patients with serum carnosinase deficiency. Homocarnosine (gamma aminobutyryl histidine) is a brain dipeptide. It is hydrolyzed by serum but not by cytosolic carnosinase. The physiological function of homocarnosine is unknown. It may act as a reservoir of GABA in some parts of the brain. The relationship between symptoms observed in serum carnosine–deficient patients and biochemical findings is uncertain.
 PROLIDASE DEFICIENCY
PROLIDASE DEFICIENCY
The hallmark biochemical finding of this disorder is massive hyperexcretion of a large number of imidodipeptides (dipeptides with an N-terminal proline or hydroxyproline particularly glycylproline) due to a deficiency of the exopeptidase prolidase (or peptidase D).
 HISTIDINE AMMONIA-LYASE DEFICIENCY
HISTIDINE AMMONIA-LYASE DEFICIENCY
Histidinemia due to histidine ammonia-lyase deficiency has been recognized for some time, since amino acid chromatography programs were performed in persons with mental retardation. It is now recognized as a probable normal variant.
 ACETYLATED AMINOACIDURIAS: AMINOACYLASE I AND II DEFICIENCY
ACETYLATED AMINOACIDURIAS: AMINOACYLASE I AND II DEFICIENCY
Aminoacylase I deficiency is responsible for a massive excretion of neutral acetyl amino acids, detected by amino acid chromatography of urine. The pathological significance of this finding is still unknown. Aminoacylase II (aspartoacylase II) deficiency is responsible for Canavan disease, a severe early infantile leukodystrophy that is biochemically characterized by a massive accumulation of acetyl aspartate in brain and body fluids, where it is detectable by gas chromatography-mass spectrometry analysis of the urines and by brain NMR spectroscopy.
 TRIMETHYLAMINURIA AND DIMETHYLGLYCINURIA
TRIMETHYLAMINURIA AND DIMETHYLGLYCINURIA
These two disorders cause the fish odor syndrome characterized by intermittent or constant excretion of volatile compounds that have an unpleasant fishy odor.1-12
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


