Optic Nerve Disorders
John Roarty
CONGENITAL ANOMALIES
 OPTIC NERVE HYPOPLASIA
OPTIC NERVE HYPOPLASIA
Optic nerve aplasia is extremely rare, and most often the diagnosis represents an extreme example of optic nerve hypoplasia. Optic nerve hypoplasia is characterized by a reduction in the number of axons within the optic nerve. The nerve head is small and often pale. There may be a white or yellow peripapillary halo surrounded by a ring of pigment (double ring sign) corresponding to the size of a normal disc (Fig. 593-1). The normal distance from the temporal disc edge to the macula is 3 to 3.4 disc diameters. If the optic nerve is small, the number of disc diameters increases to greater than 4 disc diameters. The retinal vasculature may be tortuous, and the fovea pit may be underdeveloped. On radiographic study, the optic canal is small. The diagnosis can be difficult if optic nerve hypoplasia is mild. Disc photography and quantitative assessment of optic nerve head features by a nerve head analyzer may be helpful in establishing a diagnosis.
Visual acuity ranges from normal to no light perception but is stable over time. It is difficult to predict vision outcome based on the appearance of the optic nerve head or with neuroimaging. Most cases are sporadic. Familial instances are rare. Optic nerve hypoplasia has been associated with fetal alcohol syndrome; maternal diabetes (usually a hemihypoplasia affecting the upper or lower half of the disc); maternal use of quinine, anticonvulsants, or aminopterin; maternal illicit drug use; in utero cytomegalic infection; toxemia; and adolescent pregnancy. Accompanying ocular disorders may include aniridia, albinism, coloboma, Duane retraction syndrome, high myopia, and numerous neurological and pediatric conditions.
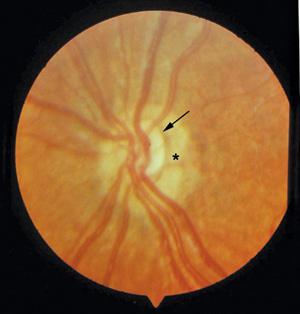
FIGURE 593-1. Optic nerve hypoplasia. Arrow points to optic nerve. Surrounding hypopigmentary area (*) makes up the “double ring” sign and represents the scleral canal through which a normal-sized optic nerve would have run.
Up to 78% of affected patients have bilateral optic nerve hypoplasia, and 75% have neurodevelopmental problems that accompany bilateral involvement. In bilateral optic nerve hypoplasia, particularly in severe forms, poor vision and nystagmus may be apparent early in life, and there may be associated abnormalities such as mental retardation, delayed development, seizures, deafness, cerebral atrophy, hemiparesis, ventricular defects, porencephalic cyst, hypopituitarism, hypothyroidism, and diabetes insipidus. Sudden death has been reported in response to a viral illness likely due to unrecognized pituitary insufficiency. Septo-optic dysplasia, also known as de Morsier syndrome, is one of the more common bilateral optic nerve hypoplasia syndromes. It is characterized by midline central nervous system abnormalities such as hypothalamic dysfunction, pituitary dysfunction, and agenesis of the septum pellucidum; malformation of the optic chiasm; and agenesis of the corpus callosum. Patients have a characteristic facies with a broad forehead and large anterior fontanelle. Mutations in the autosomal dominant HESX1 gene at 3p21.2-p21.1 may be responsible in many affected patients.1
Because of the high incidence of associated central nervous system anomalies and endocrinologic problems, all patients with optic nerve hypoplasia, particularly if bilateral, need a complete neurological workup, including brain computerized tomography (CT) or magnetic resonance imaging (MRI). An absent infundibulum of the pituitary or an ectopic posterior pituitary bright spot are characteristic MRI findings. Endocrinologic evaluation should include growth hormone, adrenal, and thyroid function. Patients with partial absence of the septum pellucidum have a threefold lower risk of pituitary hormone deficiency than those with complete absence of the septum pellucidum. Abnormalities of the hippocampus correlate with severe neurological problems. Early recognition and treatment of the hormonal imbalance permits prompt endocrinologic management and helps the child achieve normal growth and development.2
In unilateral optic nerve hypoplasia, serious central nervous system abnormalities are present in about 20% of affected individuals. Patients are typically seen because of strabismus and decreased vision. Amblyopia may develop in addition to the organic visual loss and should be treated if the visual impairment directly due to the optic nerve malformation is felt not to be profound. Because it is difficult to predict visual potential based on the appearance of the nerve, patching of the unaffected (or less affected) eye is often indicated. If the hypoplasia is unilateral or asymmetric, there may be an afferent pupillary defect (Marcus Gunn pupil).3
 COLOBOMA
COLOBOMA
Optic nerve coloboma is due to incomplete closure of the posterior end of the embryonic fissure of the optic cup in the seventh week of gestation. This process is in part driven by the PAX2 transcription gene at 10q25, mutation of which can result in the renal-coloboma syndrome characterized by renal malformations and isolated optic nerve coloboma. Most cases of coloboma are sporadic. Optic nerve coloboma may also be part of an incomplete closure of the rest of the fissure affecting the iris, or choroid (with dysplasia of the overlying retina). Multiple other genes have been implicated in this process.
The colobomatous optic nerve appears as an enlarged, white, excavated nerve head with or without coloboma of the surrounding choroid (Fig. 593-2). Microphthalmia may be present. If large, it may include the macula. Visual acuity can be very good if coloboma is isolated to the nerve. Subarachnoid cerebrospinal fluid around the optic nerve may flow through the defect under the retina, interfering with macular function.
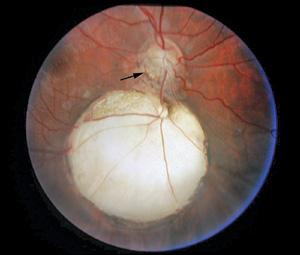
FIGURE 593-2. Optic nerve coloboma. Note inferior border of optic nerve (arrow) is incomplete, larger, and ill defined. Below the optic nerve is a large area of dysplastic retina overlying sclera with no intervening choroid surrounded by hyperpigmented area. This is a “retinal” coloboma.
 OPTIC NERVE PIT
OPTIC NERVE PIT
Optic nerve pits are a mild form of nerve head coloboma characterized by dark gray oval or slitlike holes in the disc, usually on the infer-otemporal side or centrally. They are usually unilateral and most often have no hereditary pattern. A wide variation in visual acuity can occur, and the vision is usually normal. Optic pits are important clinically, because they may produce scotomas, at times associated with serous (caused by fluid under the retina) retinal detachments.4 Although retinal detachment has been seen in children in the first decade, it occurs in up to 50% of the patients at a mean age of 31 years. It is believed that cerebrospinal fluid tracks under the retina, causing the detachment. Attempts to repair the elevated retina with silicone oil injected into the eye have resulted in the silicone oil migrating into the subarachnoid space of the optic nerve.5 Systemic abnormalities are not commonly associated with congenital optic nerve pits.
 MORNING GLORY DISC
MORNING GLORY DISC
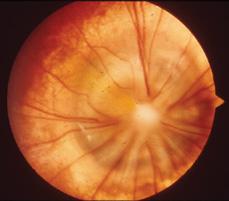
This optic disc anomaly can be considered a form of optic nerve coloboma. Most instances are unilateral and nonfamilial. The disc appearance resembles the morning glory flower. The optic nerve is located in the center of an enlarged, excavated disc with central fibroglial tissue (Fig. 593-3). It can be orange or pink in color, with the retinal vessels splayed out at the periphery of the disc. It is twice as common in females as in males, and the right eye is more frequently involved. Visual acuity is usually 20/100 or worse, although it is difficult to predict the visual potential based on the appearance of the anomaly. Patients with vision as good as 20/40 are known. Patching the unaffected eye may be helpful in reversing amblyopia, which complicates the vision loss. High myopia and visual field defects are prominent. Serous retinal detachment is the most serious ocular complication and may occur in up to 38% of the patients. Although usually isolated, association with sphenoidal encephalocele is known and may be suspected if the patient also has a notch in the center of the lip. Morning glory optic nerve has also been reported in trisomy 4q and the PHACE syndrome (posterior fossa malformations; hemangiomas; arterial abnormalities; cardiac defects, especially aortic coarctation; and eye abnormalities).6
 MYELINATED NERVE FIBERS
MYELINATED NERVE FIBERS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


