Louise Allen • Know and understand the anatomy and embryology of the eye • Understand how the structure of the eye relates to function • Understand the normal development of vision and the pathophysiology of visual impairment • Know the physiology of the eye and its movement • Know the genetic and environmental factors in the aetiology of eye disorders • Recognize congenital eye disease, enabling early prevention and treatment of blinding conditions • Know when an ophthalmic phenotype can help to make a systemic/genetic diagnosis • Know when a systemic disease puts a child at risk of ophthalmic disease
Ophthalmology
Although ophthalmology may seem a fairly minor topic to the generalist, paediatric eye and visual disorders are common in both primary and secondary care settings. The eyes, their visual pathways and higher visual processing mature throughout early childhood. Good visual function depends on all these factors. These stages of normal visual development and the way we assess them are covered in Chapter 4, Normal child development.
Epidemiology of childhood visual impairment
Visual impairment in childhood impacts all areas of a child’s development and influences their future prospects. In the UK, nearly 4% of the childhood population are registered severely visually impaired or blind (compared to 12% in developing countries) and half these children will have additional motor, sensory, learning impairments or systemic disease. Our screening programmes and access to specialist paediatric ophthalmic care prevent many cases of severe visual impairment, and the majority (75%) of children registered blind in the UK have an unpreventable and untreatable cause. The registration process facilitates the educational and social support these children need. The level of visual impairment is based on corrected binocular visual acuity (using a Snellen chart) and visual fields. The definitions of sight impairment are shown in Box 30.1.
Applied embryology of the eye
The developing eye
The eye develops from a complex, coordinated interaction between surface ectoderm, neuroectoderm and the mesenchyme (comprising mesoderm and neural crest cells), mediated by many developmental genes (Fig. 30.1). The optic fissure closes in the sixth week of gestation and, by the beginning of the second trimester, the rudimentary eye has developed, although subsequent differentiation and maturation occur beyond term.
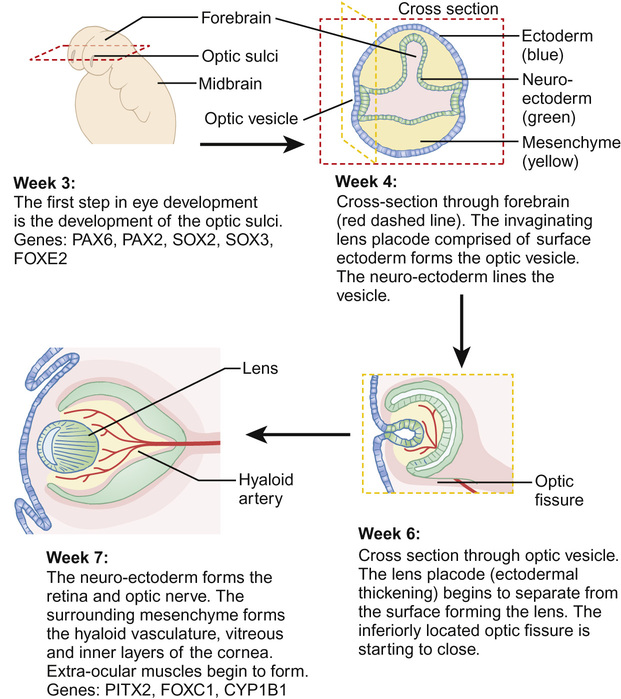
Abnormal ocular development
Anophthalmos, microphthalmos and coloboma
This is a spectrum of early ocular malformation and may be unilateral or of varying severity in each eye. The most severe manifestation is anophthalmos, where no globe is present (incidence 2/100,000 live births). In microphthalmos, a small globe, which may or may not have visual potential, is present (incidence 19/100,000 live births). An absent or very small globe will fail to stimulate orbital growth; orbital expanders and conformers should be fitted within weeks of birth in order to prevent permanent orbital asymmetry. Ocular coloboma is a more common malformation (incidence 1/20,000 live births), resulting from failure of closure of the optic fissure in the sixth gestational week. This is often an isolated anomaly but may also characterize a syndrome (e.g. CHARGE: Coloboma, Heart abnormalities, Atresia choanae, Retardation of Growth and development and Ear/hearing abnormalities). The coloboma can affect the inferonasal iris (Fig. 30.2A), and/or the inferior choroid and retina and the optic nerve, the latter two resulting in visual impairment (Fig. 30.2B).
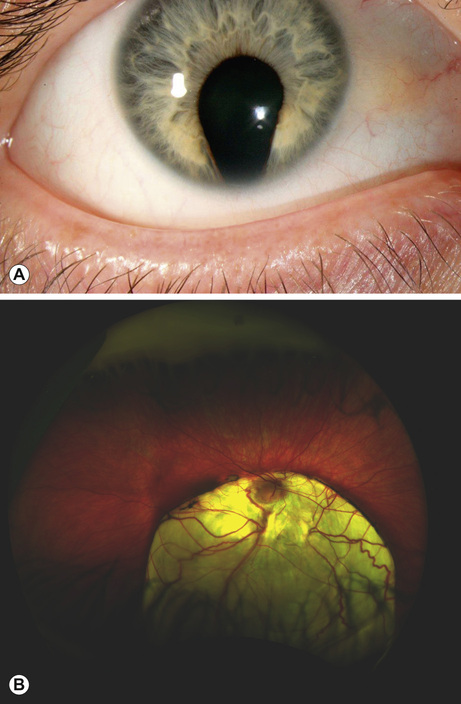
Aniridia
Although the name suggests otherwise, this is a malformation of the whole eye, usually causing partial or total absence of the iris, cataract, corneal stem cell failure and absence of the fovea (foveal hypoplasia), which causes poor vision and nystagmus. Most cases are autosomal dominant (due to inherited point mutations in PAX6). However, in sporadic aniridia, a novel micro-deletion can involve PAX6 and a neighbouring tumour suppressor gene causing WAGR syndrome (Wilm tumour, Aniridia, Genito-urinary abnormalities and Retardation): newborns with sporadic aniridia require urgent investigation for renal tumours.
Albinism
Melanin has several important functions in the developing eye. It mediates the complex organization of the fovea and guides the axons of the retinal ganglion cells distally through the optic chiasm. Children born with the more severe forms of autosomal recessive oculocutaneous albinism will have translucent irides, foveal hypoplasia and abnormal axonal decussation at the optic chiasm (a useful diagnostic feature demonstrated by visual evoked potentials). X-linked ocular albinism causes the ocular features of albinism without significant cutaneous involvement.
Vitreo-retinal dysplasia
This is thought to be caused by abnormal retinal vasculogenesis. At its most severe, the retina is an unrecognizable funnel of neurovascular tissue projecting from the optic disc into the vitreous cavity at birth. In less severe cases, there are localized areas of retinal non-perfusion and ischaemia; early recognition and laser treatment to these areas can prevent neovascularization and subsequent retinal detachment. Babies with incontinentia pigmenti (IKBKG gene mutation, X-linked dominant, lethal in males) are at risk of developing abnormal retinal vasculature and require regular retinal examination from birth in order to prevent retinal detachment with laser treatment.
Optic nerve hypoplasia
Optic nerve hypoplasia causes a small optic nerve head on fundoscopy, often associated with tortuous retinal veins and a variable level of visual impairment. Maternal risk factors include young age, maternal diabetes and a history of excessive alcohol or use of illicit drugs during pregnancy. There are many systemic associations of optic nerve hypoplasia, including fetal alcohol syndrome and Dandy–Walker syndrome. More commonly, optic nerve hypoplasia is associated with endocrine abnormalities due to panhypopituitarism (may present as neonatal jaundice, hypoglycaemia and seizures) or specific mid-line CNS malformations, such as septo-optic dysplasia, characterized by absence of the septum pellucidum and thinning or agenesis of the corpus callosum.
Applied anatomy of the eye
The developed eye (Fig. 30.3)
The axial length of the eye at birth is around 17 mm. The globe grows particularly rapidly in the first six months, reaching the adult axial length of 22 mm by 5 years. The volume of the neonatal eye is approximately 2.8 cm3 compared to the adult volume of 7 cm3. Orbital volume doubles in the first year of life and is dependent on the presence of the globe.
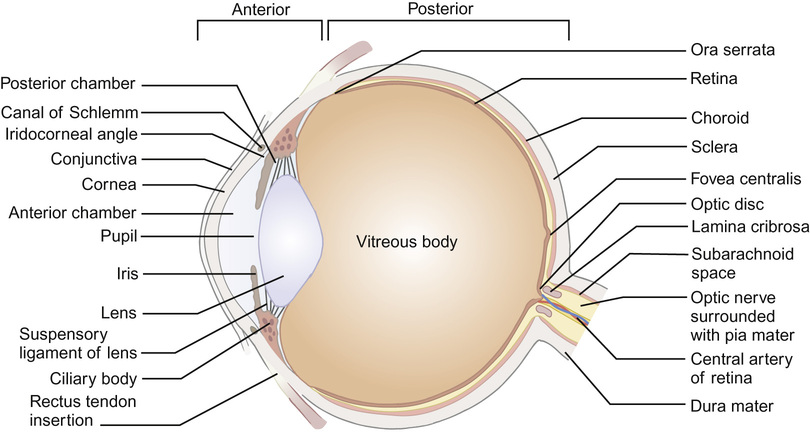
The eye is described as having an anterior segment (including conjunctiva, episclera and the externally visible portion of sclera, cornea, anterior chamber, iris and lens) and a posterior segment (including vitreous cavity, retina, retinal pigment epithelium and choroid and posterior sclera). The ophthalmic artery (a branch of the internal carotid artery) supplies the eyelids and orbit. The central retinal artery branches off the ophthalmic artery to enter the eye via the optic nerve head, supplying the inner layers of the retina (and, in the fetus, the lens via the hyaloid artery system). The posterior ciliary arteries, derived from the ophthalmic artery, anastomose within the choroid to supply the retinal pigment epithelium and outer layer of the retina, including the photoreceptors. A blood–ocular barrier exists to protect the eye from toxic substances: the blood–retina barrier is maintained by the tight junctions in the retinal capillary bed and between retinal pigment epithelial cells, and the blood–aqueous barrier by the tight junctions of the ciliary epithelial cells. Inflammation, as seen in uveitis, breaks down the blood–ocular barrier, allowing inflammatory cells and proteins to enter the aqueous and vitreous from the circulation.
The eye is also immunologically isolated (privileged): if antigens from the eye enter the systemic circulation, for example following penetrating eye injury, this can result in an autoimmune attack on the other eye – a devastating, potentially blinding condition called sympathetic uveitis.
Sensation from the orbit is conveyed via the ophthalmic division of the trigeminal nerve. Neuronal connections exist between the trigeminal nucleus and the parasympathetic dorsal nucleus of the vagus nerve. Pressure on the eye(s) during retinopathy of prematurity screening or the traction on the muscles during squint surgery can result in bradycardia and respiratory depression. Immediate cessation of the stimulus combined with administration of an antimuscarinic agent, such as atropine, can reverse the effect. Motor innervation of the orbit is discussed later in this chapter.
The structure of the retina
The retina is a complex, multi-layered neural structure lining the posterior segment of the globe. It is continuous with the optic nerve posteriorly and fuses anteriorly with the epithelium of the ciliary body at the ora serrata. The retina and retinal pigment epithelium (RPE) are derived from two neuroectodermal layers separated by a potential space. A retinal detachment occurs when fluid enters this space, peeling the retina from the underlying RPE. The retina itself is made up of many layers of neural cells: the outermost layer of the retina is made up of the photoreceptors, rods and cones, which receive metabolic and nutritional support from the underlying RPE and choroid. Central and colour vision is provided by the macular retina, the centre of which is called the fovea.
Rods are sensitive to low levels of light and are most numerous in the retinal periphery, giving peripheral and night vision. Cones are classified according to the sensitivity of their photo-pigment: long (red), medium (green) and short (blue) wavelengths. The fovea is responsible for visual acuity; cell bodies and axons of the inner retina are displaced peripherally at the fovea, making it the thinnest area of the retina and affording its tightly packed cone photoreceptors (147,000/mm2) optimal exposure to incident light. The photoreceptors are the sensory receptors of the retina, the bipolar cells, situated in the inner retina, are the first order neurons and the retinal ganglion cells (RGC), situated in the innermost layer, are the second order neurons. The RGC axons traverse the surface of the retina in the nerve fibre layer on the surface of the retina and then travel within the optic nerve to synapse in the lateral geniculate body. For optimal light transference to the underlying photoreceptors, the RGC axons are only myelinated distal to the optic disc, so that they remain transparent. Sometimes the myelination process occurs anterior to the optic disc, giving a white appearance to the nerve fibre layer; although striking in appearance, myelinated nerve fibres rarely cause visual problems unless they involve the macula.
Methods of visualizing the fundus
Although portable and cheap, the direct ophthalmoscope is the most difficult method of visualizing the fundus in children: it gives a real, magnified image but small field of view with no depth, making fundoscopy difficult in young children. In this situation, the head-mounted indirect ophthalmoscope is the instrument of choice, giving a stereoscopic, less magnified image with a much wider field of view. The virtual nature of the image, inverted both vertically and horizontally, can be confusing for the novice and the technique can take years to master. The Retcam is a wide-angle camera, which can usefully document the fundus appearance and is particularly useful for retinopathy of prematurity and retinal haemorrhages secondary to inflicted head injury. Its drawback is that the camera lens makes contact with the cornea, making it poorly tolerated in alert babies and toddlers. Most children over 5 years of age are able to sit still for standard retinal photography.
Ocular coherence tomography has been a great advance in retinal imaging, enabling a detailed cross-sectional image of the retina and optic disc to be constructed. This can be particularly helpful when macular pathology is suspected or to document changes over time, for instance, serial nerve fibre layer thickness analysis in papilloedema. Intravenous fluorescein angiography is useful for demonstrating the vascular network of the retina, particularly areas of ischaemia or vascular leakage. Once visualized by fluorescein angiography, the areas of retinal ischaemia can be lasered to prevent secondary neovascularization.
Prematurity and the eye
Retinopathy of prematurity
Vascularization of the retina begins at about 14 weeks’ gestation and is not complete until term. It is stimulated by vascular endothelial growth factor (VEGF-A) and insulin-like growth factor (IGF-1), which work synergistically. VEGF production is induced by physiological fetal retinal hypoxia, whilst IGF-1 is oxygen independent, with rising levels stimulating retinal angiogenesis during the second and third trimesters.
Retinopathy of prematurity (ROP) is a neovascular disorder affecting infants born at less than 32 weeks’ gestational age. Extremely low birth weight (<1000 g) and early supplemental oxygen requirement and acidosis are additional important risk factors. About 50% of premature babies born <1000 g birth weight will develop ROP and 15% will reach the threshold for treatment. In the UK, guidelines specify that all babies under 32 completed weeks’ gestation and less than 1500 g should be screened by an ophthalmologist.
ROP develops in two distinct phases:
The international classification of ROP includes stages of ROP development and zones of location in the retina (Fig. 30.4). Zone 1 and posterior zone 2 ROP is the most aggressive. The threshold for treatment is based on the zone of the disease, its stage, and the presence of retina vascular dilatation and tortuosity – ‘plus’ disease. Timely treatment can prevent progression to retinal detachment and blindness. Recently, a correlation between a drop-off in postnatal weight gain (which mirrors a fall in IGF-1 levels) and the subsequent development of ROP has been identified. A computer-based algorithm based on postnatal weight gain (WINROP) is being developed, which may help future identification of those babies most at risk.
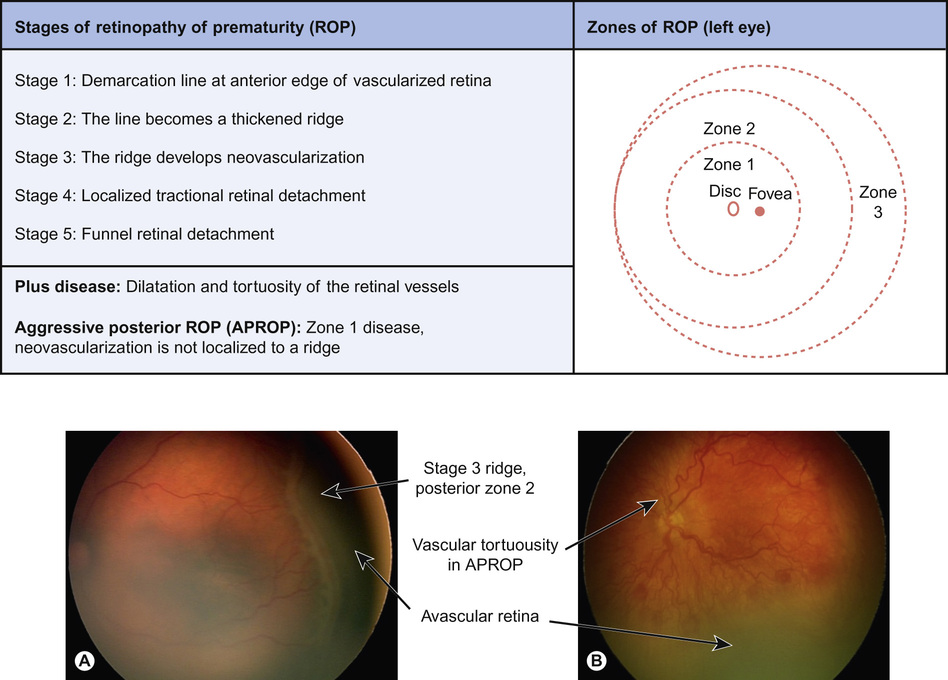
Currently, laser ablation of the avascular, ischaemic anterior retina is the preferred treatment for severe ROP. A promising new treatment is the intra-vitreal injection of an anti-VEGF agent (bevacizumab or ranibizumab). Although this treatment is easier and quicker to perform than laser, systemic absorption depresses serum VEGF levels for several weeks, which may have systemic consequences. The resultant prolonged absence of VEGF within the eye also delays the normal vascularization of the anterior retina, leaving it non-perfused and ischaemic for months beyond term. In the UK, anti-VEGF treatment is currently reserved for the most severe cases in which laser treatment is not possible or sufficiently effective. Randomized, controlled trials are planned to determine the safety of this new treatment.
Congenital and developmental eye conditions
Childhood cataract
Cataracts may be present at birth or develop and become visually significant with time. The lens grows in size throughout life as layer upon layer of lens fibres are laid down, encircling the fetal and embryonic nucleus like layers of an onion. The lens has the highest protein content of any tissue in the body and is transparent because of the accurate organization of the proteins (called crystallins) in fibres within it; disorganized protein fibre structure or the accumulation of abnormal metabolic products within the lens causes opacification.
Congenital cataract
Cataracts occur in 2/10,000 live births and are usually identified at screening. Although the majority are idiopathic, 20% of children with isolated bilateral congenital cataracts will have a family history or parental consanguinity; the cataracts are often secondary to point mutations (in genes such as MAF, CRYA1). Cataracts are common features of many different syndromes, particularly Down’s syndrome (1.5% prevalence); they may also complicate other ocular malformations, such as aniridia. Babies with sporadic bilateral congenital cataracts should have a TORCH and galactosaemia screen and a referral for genetic evaluation. Males should additionally have a urinary amino acid assessment to exclude Lowe syndrome.
Unilateral congenital cataracts most commonly result from abnormal regression of the embryological hyaloid vascular system, which supplies the posterior lens during development (persistent fetal vasculature). Unilateral cataracts are usually associated with mild microphthalmia and are not generally investigated.
Cataracts developing after the critical period of neuroplasticity (see Chapter 4, Normal child development) are usually associated with a better visual prognosis. These may be genetic in aetiology but may also be secondary to uveitis, steroid therapy and radiation.
Management of childhood cataract
Visually significant congenital cataracts require early surgery within the first two months of life to achieve good visual function. Less dense cataracts may be managed conservatively with close observation and refractive correction. If the cataracts are unilateral or asymmetrical, occlusion therapy (patching) of the better-seeing eye may be prescribed.
Infants and children develop especially severe ocular inflammation and fibrotic changes following intra-ocular procedures. To prevent re-opacification of the visual axis with scar tissue, the posterior capsule and anterior vitreous are removed during the lensectomy procedure and an intensive topical steroid regimen is prescribed postoperatively. Some paediatric ophthalmologists elect to implant the eye with an acrylic intra-ocular lens (IOL) at the time of lensectomy if the globe is otherwise normal; others leave the eye aphakic (without a lens) and replace the refracting power of the lens with a contact lens until the child is older. Since they are not exchangeable, an IOL refractive power is selected to leave the eye hypermetropic, to allow for the physiological myopic shift which occurs during ocular growth. This residual hypermetropia is usually corrected with an extended wear contact lens for the first year and glasses thereafter.
Childhood glaucoma
Childhood glaucoma is a rare, potentially blinding condition characterized by raised intra-ocular pressure and optic disc cupping. In the normal optic nerve head, the retinal ganglion cell (RGC) axons are concentrated around the circumference of the optic disc, leaving a pale central area relatively devoid of axons, called the optic cup. The raised intra-ocular pressure of glaucoma causes RGC death and, as the number of axons passing through the optic nerve head opening dwindles, the relative size of the optic cup (the cup : disc ratio) gradually enlarges. Uncontrolled glaucoma will result in peripheral visual field loss, which is difficult to detect in children. The RGCs serving the macula are the last to be damaged; this is why the optic neuropathy associated with glaucoma is not characterized by the early loss of visual acuity or colour vision that is seen with optic neuritis or optic atrophy.
The normal intra-ocular pressure in children is usually between 6 and 18 mmHg and can usually be measured in the eye clinic. Raised intra-ocular pressure is the result of impaired aqueous outflow through the trabecular meshwork rather than overproduction of aqueous by the ciliary body. Children under 3 years of age have low scleral rigidity, and raised intra-ocular pressure will result in globe expansion (buphthalmos); the increase in axial length causes a shift towards myopia (or loss of hypermetropia) and an increasing corneal diameter (normal corneal diameter is 11 mm).
Primary congenital glaucoma
Presenting within the first year of life, this is usually bilateral, and results from abnormal development of the drainage angle. Incidence is 1/10,000 live births and the majority are due to a gene mutation (in the CYP1B1 gene). The globe enlarges due to the raised pressure and splits occur in the deeper layers of the cornea (Haab’s striae) leading to photophobia and corneal opacification. Treatment is by the early surgical division of the abnormal ‘Barkan membrane’ which obstructs fluid flow to the drainage angle.



