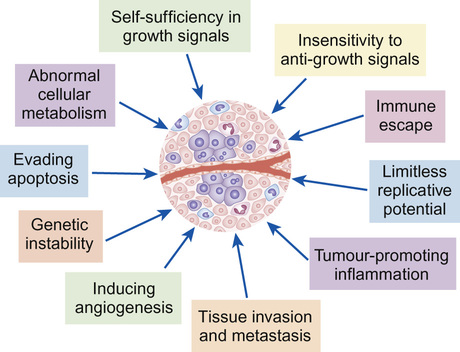
Daniel Morgenstern, Rachel Dommett Most simply, cancer can be described as uncontrolled cellular proliferation. Most healthy cells have a limited potential for replication and their proliferation, differentiation and death is carefully controlled. During cancer development, cells undergo a multistep process in which they gradually acquire genetic mutations allowing them to escape these normal controls. Benign, or low-grade, tumours tend to grow locally and have not developed the potential to metastasize (spread to other tissues). By contrast, high-grade, or malignant, tumours are highly proliferative (have a high ‘mitotic index’ when examined under the microscope) and tend to metastasize. However, a benign tumour does not necessarily equate with a benign clinical course, particularly if the tumour is growing close to and impinging on a vital structure (such as a low-grade glioma growing within the optic pathway which, untreated, may lead to blindness). The biology of cancer is complex. However, although the details vary between cancer types, the underlying principles are similar. Normal cells and tissues have mechanisms designed to prevent cancer development and a cell must overcome these in order to become malignant. The key features of cancer development are shown in Figure 22.1. Cancer occurs when normal protective mechanisms are lost. Cancer develops when cells develop one, or more, of the following: Genetic abnormalities in cancers can be divided into those that result in over-activation of a particular gene or pathway and those that result in loss of function. Gain of function of (proto-) oncogenes, such as a growth factor receptor signalling pathway or transcription factor, enhances malignant progression (Fig. 22.2). Conversely, the loss of function of tumour suppressor genes, such as those that trigger apoptosis or block cell cycle progression in the presence of DNA damage, also drives cancer development. Cancer is rare in childhood and accounts for less than 1% of all cancers. In the western world around 1 in 500 children will develop some form of cancer by 14 years of age. In the UK approximately 1600 children are diagnosed with cancer each year. The 12 major diagnostic groups (and most common tumour types in children) are: leukaemias; lymphomas; brain and spinal tumours; sympathetic nervous system tumours (neuroblastoma); kidney tumours (Wilms’ tumour); soft tissue sarcomas (rhabdomyosarcoma); bone tumours (osteosarcoma, Ewing’s sarcoma); liver tumours (hepatoblastoma); retinoblastoma; gonadal and germ cell tumours; epithelial tumours (carcinomas); and others. The incidence of different tumours varies dramatically with age (Fig. 22.3). In infants, the most common solid malignancies are embryonal tumours such as neuroblastoma, retinoblastoma and hepatoblastoma. The incidence of acute lymphoblastic leukaemia (ALL), the most common type of leukaemia seen in childhood (and the most common childhood malignancy overall), is highest among children aged 2–3 years. Brain and spinal tumours are, as a group, the most common solid tumours seen in children. By contrast, lymphoma, particularly Hodgkin’s lymphoma, bone tumours and malignant melanoma are rare in early childhood, but show a marked increase in incidence in adolescence. Gonadal germ cell tumours display a bimodal distribution in boys with a peak in infancy and early childhood, and their incidence rises again in teenagers. Carcinomas are rare in children, but become progressively more common in adolescents and younger adults. Overall (for reasons not fully understood), childhood cancer is about one-fifth more common among boys than girls. Outcomes have improved dramatically over the last 40 years due to the introduction of chemotherapy and other treatment modalities, and collaborative national and international clinical trials (Fig. 22.4). Paediatric oncology has led the way in treating patients in clinical trials; this has allowed new treatment approaches to be developed and tested. Children are managed according to well-defined treatment protocols that have undergone frequent revision as more has been learnt about the best way to treat particular cancer types. Overall, childhood cancers are rare and clinical trials typically require national and/or international collaboration in order to recruit sufficient numbers of patients. Clinical trials for childhood ALL have typically been UK-based, whilst those for rarer solid tumours (such as neuroblastoma, rhabdomyosarcoma or hepatoblastoma) have only succeeded because of international collaboration. Most of the historical improvement in outcomes has come about from the development of standardized protocols using a combination of cytotoxic chemotherapy agents plus (where appropriate) surgery and/or radiotherapy. As our understanding of the molecular basis of childhood cancer has improved, this has led to better risk stratification and adapted therapy. The hope for the future is that it will also be possible to target these molecular abnormalities to provide specific targeted therapies. Significant improvements have been made in the outcomes for childhood cancer patients over the last 30–40 years (see Recent advances in science that have changed clinical practice), largely as a result of collaborative clinical trials. Newly diagnosed patients will be treated as part of a Phase III clinical trial or according to an established treatment protocol where no trial is currently open. These trials will typically seek to compare two established treatment regimens in a randomized study. By contrast, new drugs (such as new molecularly-targeted agents) are generally explored at a limited number of centres and typically these studies recruit patients with relapsed or refractory disease for whom there are no standard treatment options. Initial Phase I studies are designed to assess toxicity and gather information about drug metabolism (pharmacokinetics). They may also include pharmacodynamic analyses that are designed to demonstrate the effect of the drug on its target (e.g. inhibition of a particular enzyme). Subsequent Phase II studies then focus on obtaining evidence of efficacy against a particular disease or diseases. New drugs that are particularly promising may then be taken forward to Phase III studies and may ultimately become incorporated into standard upfront therapy. The ways in which clinical trials have helped improve outcomes of childhood cancer is considered further in Box 37.9. Although there is much in common between adult and paediatric cancers, there are also fundamental differences in how they arise. The most common adult cancers (breast, colon, prostate and lung) are carcinomas, i.e. a cancer derived from epithelial cells, and typically result from acquired chronic cellular insults, for example from tobacco smoke. By contrast, the most common paediatric solid tumours (brain tumours, neuroblastoma, Wilms’ tumour and rhabdomyosarcoma) typically arise in young children who have not had such exposures. Childhood cancers tend to derive from primitive, poorly differentiated cell types and likely represent an abnormality arising from fetal development. Indeed, there are striking parallels between cellular behaviour during fetal development and malignancy, such as proliferation, invasion and cellular migration to new tissues. In many cases, a greater understanding of the normal processes of embryological development has aided our understanding of how paediatric cancers arise. The precise cause of most childhood cancers remains unclear but is likely to involve an interaction between environmental factors (e.g. viral infection) and host genetic susceptibility. Published evidence confirms that there is a small increased risk of malignancy associated with radiation exposure from medical imaging such as CT scanning, although this is unlikely to be a significant contributory factor in most instances. Worldwide, the most important examples of childhood cancers caused by infections are Burkitt’s lymphoma, Hodgkin’s lymphoma and nasopharyngeal carcinoma (all associated with Epstein–Barr virus), hepatocellular carcinoma (hepatitis B) and Kaposi sarcoma (HHV8). However, overall these associations account for a very small proportion of childhood cancer in western countries. The early childhood peak of leukaemia incidence in affluent western populations and the persistently lower incidence in socio-economically disadvantaged groups and less developed countries has suggested that acute lymphatic leukaemia could be associated with an abnormal response to a common infectious agent. Parental smoking during pregnancy, or during the period prior to conception, has been implicated as a cause of hepatoblastoma and a meta-analysis has shown a 10% increase in risk of all cancers with maternal smoking during pregnancy. Overall, environmental risk factors have not been established for the vast majority of childhood cancers. The risk of developing leukaemia in Down’s syndrome is increased 30-fold compared with the general population. Cancer predisposition syndromes have provided vital clues to the underlying genetics of cancers, however they account for less than 5% of cases. This is well illustrated in hereditary retinoblastoma, which provides a model for understanding the principles of inherited predisposition, as it is linked to a single gene. Familial retinoblastoma accounts for approximately 40% of cases. Presentation is usually early (first year of life) and bilateral. Sporadic cases present later, are unilateral and are not associated with a positive family history. Survivors of familial retinoblastoma have a very high risk of developing second primary tumours, of which osteosarcoma is the most common. These features of retinoblastoma were noted by Knudson and led him to propose the ‘two-hit’ mutational hypothesis. This states that two mutations are necessary in a cell for a tumour to develop. In hereditary cases, the first mutation is germline, while the second is somatic. In sporadic cases, two somatic mutations are required in the same cell for a tumour to develop. Knudson’s hypothesis was confirmed in the 1980s with the identification of the retinoblastoma tumour suppressor gene (RB1). Retinoblastoma results as a consequence of two mutations in the RB1 gene within the somatic cells of the retina. In comparison to retinoblastoma, familial clusters of other embryonal tumours are rare. For example, in Wilms’ tumour, inherited cases are thought to represent only 1% of all cases. Beckwith–Wiedemann is the most common syndrome associated with Wilms’ tumour. Organomegaly, macroglossia, hemihypertrophy, neonatal hypoglycaemia and exomphalos are all features of this fetal overgrowth syndrome. Around 10% of cases develop tumours, of which Wilms’ is the most common, and imprinting of genes on chromosome 11 has been implicated as a causative mechanism. Other syndromes associated with Wilms’ tumour include Denys–Drash, WAGR (Wilms’ tumour, aniridia, genitourinary tract abnormalities and mental retardation) and isolated hemihypertrophy. Neurofibromatosis type 1 (NF1) is inherited as an autosomal dominant condition, although many cases are new mutations. NF1 is associated with an increased risk of brain and spinal cord tumours, particularly low-grade gliomas of the optic pathway. Although familial causes for childhood cancer are infrequent, a thorough family history is very important when assessing any new case of childhood cancer and referral to a geneticist for further assessment may be appropriate. In patients with a strong family history (particularly of breast cancer, brain tumours or sarcoma), a diagnosis of Li–Fraumeni syndrome should be considered. This cancer predisposition syndrome results from mutation and loss of function of the p53 tumour suppressor gene and may be inherited, or occur de novo in a cancer patient. Knowledge of a cancer predisposition syndrome (e.g. NF1, familial retinoblastoma) is important when planning treatment, as it may be necessary to avoid certain treatment modalities (e.g. radiotherapy) to reduce the risk of secondary malignancies. Survivors of childhood cancer have an increased risk of developing a subsequent malignancy. This may reflect a genetic predisposition but may also be due to the treatment they received for their first cancer. Radiotherapy is associated with second malignancies that may arise many years later, particularly in parts of the radiation field that received lower doses of radiation that damage but do not kill cells. Breast cancer in patients who received thoracic radiotherapy for Hodgkin’s lymphoma is a particular risk. Chemotherapy drugs (such as cyclophosphamide and etoposide) are associated with a small increased risk of secondary leukaemia. Early symptoms of cancer in children are often non-specific and easily explained by more common illnesses, which can lead to delays in diagnosis. Repeat attendance without a unified diagnosis warrants further investigation or referral. The history should include a developmental review, as loss of previously attained developmental milestones in a young child and changes in school performance in an older child may be the only indicators of spinal cord compression or an underlying brain tumour. Common patterns of presentation are shown in Table 22.1. Table 22.1 Common patterns of presentation in children with malignancy Occurs due to displacement of marrow by leukaemia or disseminated malignancy. These may reflect bone marrow infiltration with leukaemia, metastases, spinal tumour or impending/actual spinal cord compression • Association with general malaise, e.g. neuroblastoma • Hypertension (secondary to compression of renal vasculature in Wilms’ tumour or neuroblastoma) N.B. May be painless and isolated finding, e.g. Wilms’ tumour
Oncology
What is cancer?
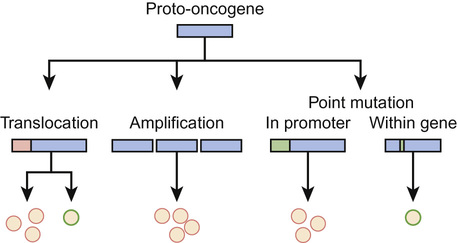
Oncogenes and tumour suppressor genes
Epidemiology and aetiology – who gets cancer?
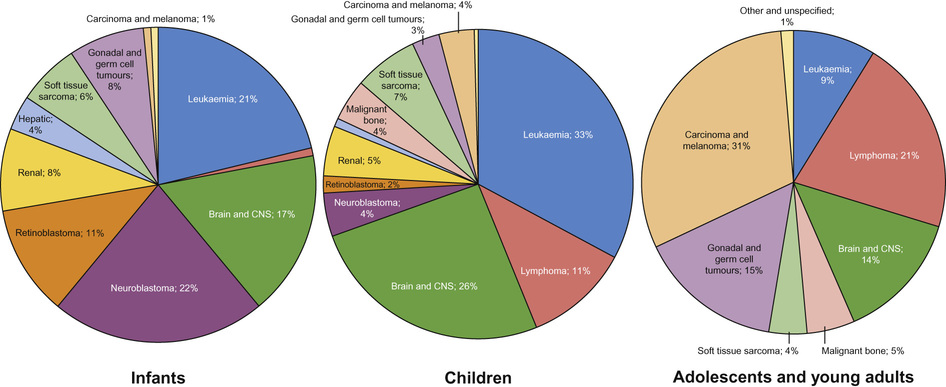
Incidence
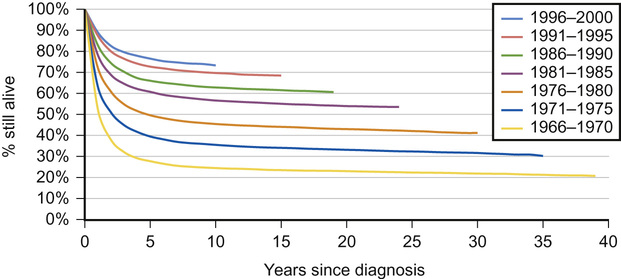
Why have survival rates for childhood cancer improved over the last 40 years?
What is the role of clinical trials in paediatric oncology?
Aetiology
Potential environmental risk factors
Radiation
Infection
Other environmental triggers
Known genetic risk factors
Cancer risk in survivors of childhood cancer
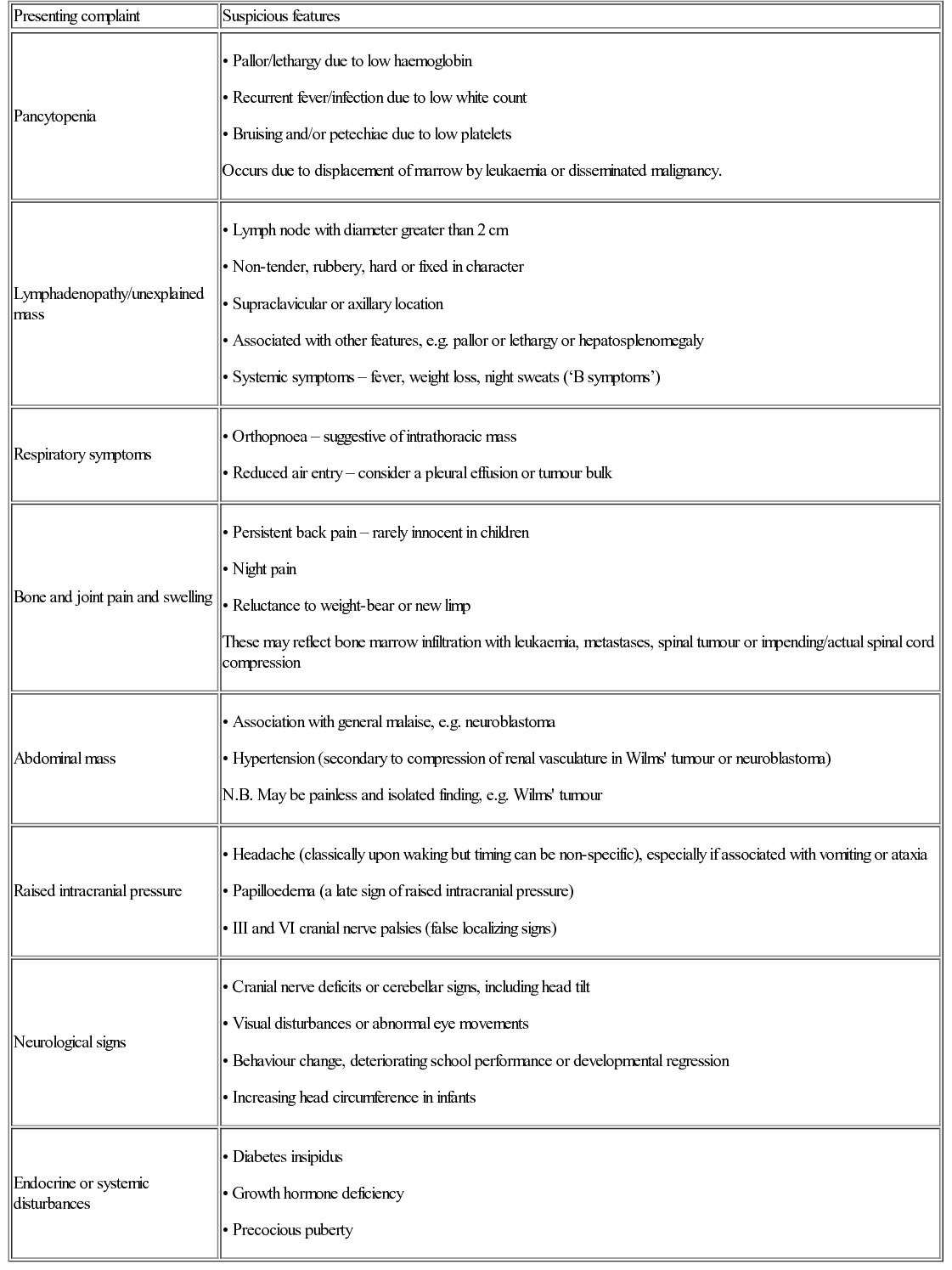
Patterns of presentation
Presenting complaint
Suspicious features
Pancytopenia
Lymphadenopathy/unexplained mass
Respiratory symptoms
Bone and joint pain and swelling
Abdominal mass
Raised intracranial pressure
Neurological signs
Endocrine or systemic disturbances
Paraneoplastic phenomena
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Oncology
Chapter 22
Learning objectives
By the end of this chapter the reader should: