Normal and Abnormal Hemostasis
Janna M. Journeycake, Janet Yang, and Anthony K. C. Chan
The hemostatic mechanism is a dynamic system that maintains the fluidity of blood, while allowing for the formation of blood clots to prevent bleeding subsequent to blood vessel injury. These interrelated components consist of vessel walls (including endothelial cells), platelets, coagulation factors, inhibitors, and the fibrinolytic mechanism. Perturbation of any of these systems can lead to disease states that are thrombotic or hemorrhagic in nature. This chapter discusses the normal and abnormal hemostatic system in children, with emphasis on disorders involving the endothelium, platelets, and defects in coagulation and fibrinolysis.
NORMAL HEMOSTASIS
 ROLE OF THE ENDOTHELIUM
ROLE OF THE ENDOTHELIUM
The endothelium is comprised of a single layer of endothelial cells lining the inner lumen of the vascular wall and the underlying basement membrane matrix that provides structural and functional support to the entire vascular network. The primary role of the endothelial cells is to act as a selective barrier between the circulating blood and the underlying tissues, ensuring vascular patency by providing a nonthrombogenic surface for circulating blood under normal, physiologic conditions. In addition, the endothelium interacts with the underlying matrix by directing the passage of molecules, nutrients, solutes, and hormones from the blood. Intact endothelial cells inhibit platelet adhesion by production of nitric oxide (NO) and prostaglandin I2 (PGI2), which also has a vasodilatory effect. Weibel-Palade bodies in endothelial cells are important for storage and sequestration of the largest multimers of von Willebrand factor (vWF).9,10 Endothelial cells also produce tissue factors and tissue factor pathway inhibitor (TFPI), tissue plasminogen activator (t-PA), prostacyclin, antithrombin (AT), and thrombomodulin, as well as the surface protein for activation of protein C (PC). 
 ROLE OF PLATELETS
ROLE OF PLATELETS
Platelets are anucleated fragments derived from megakaryocytes that are normally discoid shaped. Platelets are recruited to the site of injury, where they interact with the endothelial cells and underlying basement membrane (BM) matrix.22-24 Their role is to provide primary hemostasis, which consists of three major events. (1) When the integrity of blood vessels is broken, circulating platelets will be exposed to and interact with the adhesion molecules of the subendothelial layers, including fibronectin (via glycoprotein [GP] Ic/IIa), collagen (via platelet GP Ia/IIa) and von Willebrand factor (vWF) (via GP Ib).  (2) Second, platelet activation occurs after platelet adhesion and intra-cellular signaling. Platelets can also be activated by agonists that are released or generated at the site of injury, including thrombin, adenosine diphosphate (ADP), serotonin, and thromboxane A2. Following activation, platelets become more spherical with pseudopod extension, activation of GP IIb/IIIa, platelet secretion of granule contents containing platelet agonists and other essential molecules for hemostasis, and increased recruitment of additional platelets to the site of injury. The procoagulant anionic phospholipid molecules move to the outer surface of the plasma membrane, which enhances the anchoring of coagulation factors and facilitates secondary hemostasis. (3) Platelet aggregation, as a result of bridge formation between GP IIb/IIIa receptors on adjacent platelets, provides a scaffold for secondary hemostasis. The initiation and propagation of coagulation will eventually lead to the formation of a platelet plug (thrombus).25
(2) Second, platelet activation occurs after platelet adhesion and intra-cellular signaling. Platelets can also be activated by agonists that are released or generated at the site of injury, including thrombin, adenosine diphosphate (ADP), serotonin, and thromboxane A2. Following activation, platelets become more spherical with pseudopod extension, activation of GP IIb/IIIa, platelet secretion of granule contents containing platelet agonists and other essential molecules for hemostasis, and increased recruitment of additional platelets to the site of injury. The procoagulant anionic phospholipid molecules move to the outer surface of the plasma membrane, which enhances the anchoring of coagulation factors and facilitates secondary hemostasis. (3) Platelet aggregation, as a result of bridge formation between GP IIb/IIIa receptors on adjacent platelets, provides a scaffold for secondary hemostasis. The initiation and propagation of coagulation will eventually lead to the formation of a platelet plug (thrombus).25
Platelets are also involved in the recruitment of inflammatory cells (monocytes and neutrophils) to the site of vascular injury, release of growth factors, and stimulation of migration and proliferation of vascular smooth muscle cells and fibroblasts. 
 COAGULATION AND FIBRINOLYTIC SYSTEMS
COAGULATION AND FIBRINOLYTIC SYSTEMS
When there is vascular injury, coagulant and fibrinolytic components of the hemostatic system, in the presence of cell surfaces (endothelial cells and platelets), work together to generate a series of cascades that ultimately results in the generation of a coagulant enzyme called thrombin. Thrombin is pivotal in coagulation, with a feedback mechanism that ensures rapid thrombin generation leading to fibrin clot formation, thus preventing excessive blood loss. Once a fibrin clot is formed, the fibrinolytic system lyses the clot by promoting the generation of plasmin, a key enzyme that breaks down fibrin in the clot to restore vascular patency. In both cases, a system of checks and balances, via direct and indirect inhibitors of the key enzymes, ensures that clots and bleeds do not go uncontrolled.
The hemostatic system is continuously evolving from birth to adulthood. There are age-related differences in the composition of the hemostatic system between neonates and children, and adults. The work of Andrew et al. paved the road for research into the differences in hemostatic parameters between children and adults, establishing clinical reference ranges for fetuses, infants (pre- and full-term), and children (ages 1–16 years)29-32 (Table 435-1).
Plasma concentrations of procoagulant proteins, including vitamin K–dependent coagulation factors (FVII, FIX, FX, and thrombin) and contact factors (FXI, FXII, prekallikrein, and high-molecular-weight kininogen) are half the adult values in healthy newborns, contributing to slightly prolonged prothrombin time (PT), international normalized ratio (INR), and activated partial thromboplastin time (aPTT).29-31,33,34 The plasma concentrations of these procoagulant proteins increase to ∼80% of adult values by 6 months of life and remain somewhat decreased throughout childhood until the late teen years.32,35 Levels of some coagulation factors (fibrinogen, FV, FVIII, and FXIII) in newborns are comparable to those of adults and remain so throughout childhood.29,30,35 However, von Willebrand factor (vWF) plasma levels are elevated in fetuses36 and are twice those of adult levels in newborns,30-32 but gradually decrease over the first 6 months of life. Levels of direct thrombin inhibitors such as antithrombin (AT) and heparin cofactor II (HCII) and indirect thrombin inhibitors such as protein C (PC) and protein S (PS) at birth are also lower than in adults; levels continue to rise throughout infancy and childhood.32,35 Like vWF, plasma levels of thrombin inhibitors like α2-macroglobulin (direct)37-39 and thrombomodulin (indirect)14,32,40 are elevated at birth compared to adult levels and gradually decrease during. In contrast, PC and PS plasma levels are significantly lower at birth and eventually reach adult levels by adolescence and 3 months of age, respectively.29-32,35,41
Table 435-1. Age-Adjusted Values: Blood Coagulation Measurements Compared to Adult Norms
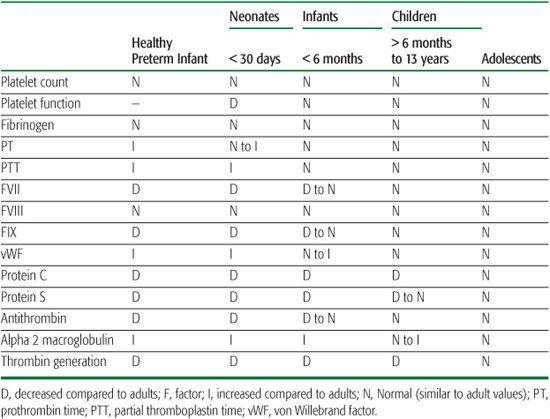
Fibrinolysis, like the coagulation system, has components that are age dependent,29,30 such as newborn plasma levels of plasminogen, a precursor to plasmin, being 50% to 75% of adult values and the primary plasmin inhibitor, α2-antiplasmin (α2 AP), ∼80% of adult values.29-31
 BLOOD FLOW AND SHEAR FORCE
BLOOD FLOW AND SHEAR FORCE
The hemostatic system has the ability to control bleeding in different size blood vessels as well as in pathologic states, where great variation in the velocity of blood flow causes different shear force acting on the platelets and the binding of clotting factors, and, ultimately, affects the formation and stability of the fibrin clot on the injured blood vessel. 
In high shear rate conditions such as the arterial circulation, the platelet must resist the force of blood flow in order to attach to the vascular subendothelium and form stable aggregates. The initial mechanisms for platelet adhesion under high shear stress are dependent on GP Ib-IX– von Willebrand factor (vWF) interactions. 
In areas of normal circulation with low shear stress, formation of reversible platelet aggregates depends on fibrinogen interaction with GP IIb-IIIa. Platelet adhesion is less dependent on GP Ib-IX. The platelets adhere through GP Ic/IIa to fibronectin and through GP Ia/IIa and GP VI to collagen. Further platelet binding of GP IIb-IIIa and fibrinogen occur only after platelet activation and in the presence of exogenous agonist.57
ABNORMAL HEMOSTASIS
Any perturbation of the balance of the coagulation system can lead to thrombosis or a hemorrhagic tendency. In the young, symptomatic thrombotic disease is rare when compared to adults, but when it does occur the outcome can be devastating, resulting in mortality or lifelong sequelae. There are myriad underlying causes and risk factors associated with thrombotic or hemorrhagic events in children.
 PATHOLOGIES ASSOCIATED WITH THE ENDOTHELIUM
PATHOLOGIES ASSOCIATED WITH THE ENDOTHELIUM
Osler-Weber-Rendu Syndrome
The Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia (HHT), is an autosomal-dominant disorder characterized by abnormal development of the vasculature, including skin and mucosal telangiectases, arteriovenous malformations (AVMs) in various internal organs, epistaxis, and aneurysms. Affected individuals have a tendency for bleeding, and clinical manifestations can present at any age with varying degrees of severity from frequent nosebleeds58,59 to hemorrhage or other complications involving the gastrointestinal (GI) tract,60 lungs,61 kidney,62 spleen,63 bladder,64,65 or liver,66 to brain.67 One study in a pediatric center found visceral AVMs and mucosal telangiectases in children with HHT that can potentially lead to life-threatening events.68 Generally, prognosis of the disease is favorable as long as bleeding is promptly recognized and adequately treated.
This genetic disorder has been associated with mutation of the endoglin gene (HHT1)69-71 or of the activin receptorlike kinase 1 (alk1) gene (HHT2).72,73
Ehlers-Danlos Syndrome
Ehlers-Danlos syndrome (EDS) is a rare genetic disorder characterized by a defect in collagen synthesis. Inheritance pattern is varied, but most types of EDS are autosomal dominant. There are a variety of problems associated with EDS, ranging from mild to life threatening, depending on the individual gene mutation. Various types of EDS have been classified, depending on the clinical manifestations, genetic transmission, and biochemical abnormality associated with this disease.74-77 Classic EDS (types I and II) symptoms include soft, highly elastic, velvety skin that is prone to tearing, bruising, or scarring with slow healing times. Joint disease and cardiac complications have also been associated with types I and II. Hypermobility (type III) affects the ligaments, leading to unstable, flexible joints that often result in painful dislocation and subluxation. Collagen provides the strength in ligaments, and due to improper collagen synthesis, the ligaments become overly stretchable, and the resultant pain is a major complication.
Vascular EDS (type IV) is the most serious type of EDS as a result of an autosomal-dominant impairment in the collagen that provides strength to vessel walls.78-80 Vascular EDS is characterized by fragile blood vessel walls and organ membranes that have a tendency to rupture or develop aneurysms. The generalized vascular fragility can cause bleeding at any site of the body. It can present with abdominal pain, hematuria, hemoptysis, retroperitoneal bleeding, muscle swelling, shock, and sudden death due to spontaneous arterial rupture, which most commonly occurred in the third to fourth decade.
Coagulation disorders and Silverman syndrome are in the differential diagnoses of childhood EDS.79 Management consists of preventive measures because there is no specific treatment. Children are advised to wear protective pads over the forehead and limbs to avoid injury. Any cutaneous sutures should be applied twice as long and deep as usual.  The symptoms of each type of EDS are not mutually exclusive because of overlapping from one type to another. Other forms of EDS exist, and more information is provided in Chapter 181 on connective tissue dysplasias.
The symptoms of each type of EDS are not mutually exclusive because of overlapping from one type to another. Other forms of EDS exist, and more information is provided in Chapter 181 on connective tissue dysplasias.
Vitamin Deficiencies
Vitamins are vital to human health, so deficiencies in certain vitamins can lead to disease states. Vitamin K plays a major role in the normal functioning of hemostasis and is essential in the production of certain coagulation proteins (prothrombin [FII], FVII, FIX, FX, protein C [PC], and protein S [PS]).81,82 The lack of adequate vitamin K can predispose to both hemorrhage and coagulopathy. Vitamin K–dependent coagulation factors of newborns are reduced to 50% of adult values; hence, prophylactic vitamin K doses are given after birth. Folate and vitamin B12 deficiency can lead to hyperhomocysteinemia, an established risk factor for cardiovascular disease and atherosclerosis (see Chapter 437).83,84 Vitamin E deficiency has been associated with the hypercoagulability of neonatal blood and may be a contributory factor in disseminated intravascular coagulation frequently experienced by newborn infants.85 A well-known condition resulting from vitamin C deficiency is scurvy. Vitamin C is required for the normal synthesis of collagen in humans, and insufficient intake can lead to formation of spots on the skin, spongy gums, and bleeding from mucosal membranes. Advanced scurvy causes open, suppurating wounds and loss of teeth. Although rare in the present day and easily treated with vitamin C supplementation, scurvy still exists in developing countries.86,87 Scurvy also causes bruises, fractures, and sores that do not heal; thus, it is sometimes mistaken for child abuse.88,89 More information on vitamin-responsive disorders is found in Chapters 147–149.
 PLATELET DISORDERS
PLATELET DISORDERS
Platelets play a role primarily in controlling hemostasis. The clinical manifestations of platelet disorders vary from minor bleeding (purpura, epistaxis, menorrhagia) to organ- or life-threatening hemorrhage. Due to its role in hemostasis, hyperreactive platelets can result in thrombotic episodes. Platelet disorders are fully discussed in Chapter 439. 
Acquired platelet pathology is most commonly caused by antiplatelet drugs. Aspirin affects platelet aggregation by blocking cyclooxygenase 1 (COX-1) activity and thromboxane A2 synthesis. These effects on platelet aggregation are permanent, whereas nonaspirin nonsteroidal anti-inflammatory drugs (NSAIDs) that also inhibit COX-1 activity exhibit alterations that are reversible.106 Some children on valproate therapy develop platelet dysfunction, thrombocytopenia, and other coagulation disturbances.107
Platelet disorders can also result in thrombosis. Atherosclerotic plaques are a result of platelet formation on the inner lumen of the endothelium. This provides a prothrombotic surface for further platelet thrombi formation and could potentially lead to arterial occlusion and ischemic injury, including stroke.108,109 Factors that contribute to a prothrombotic state, such as thrombocytosis and increased platelet aggregability, may be associated with an increased risk of ischemic stroke.110-112 Platelet disorders are discussed in Chapter 439.
 DISORDERS OF THE COAGULATION SYSTEM
DISORDERS OF THE COAGULATION SYSTEM 
Hemophilias A and B
Hemophilia A (FVIII deficiency) and hemophilia B (FIX deficiency or Christmas disease) are both X-linked recessive disorders.126 Mutations in the FVIII and FIX genes are varied and include deletions, insertions, missense, nonsense, splice site mutations, and intron 1 and 22 inversions of the FVIII gene.127 The diagnosis and management of hemophilia is discussed in Chapter 436.
The diagnosis and management of hemophilia is discussed in Chapter 436.
von Willebrand Disease
von Willebrand disease (vWD) is the most prevalent hereditary bleeding disorder, affecting up to 1% of the general population. It is classified into different types, depending on the quantitative and/or qualitative type of defect.  Further details regarding the diagnosis and management of vWD care are provided in Chapter 436.
Further details regarding the diagnosis and management of vWD care are provided in Chapter 436.
Rare Coagulation Factor Deficiencies
The factors involved in the coagulation cascade ensures clot formation in the event of vascular trauma to prevent unwanted bleeding. Abnormalities either in the quantity or function of any factors involved in coagulation can lead to clotting disorders. Most of the less common factor deficiencies are inherited autosomal-recessive disorders that manifest as bleeding tendencies. The prevalence of these disorders is 1 to 2 per million individuals.133 They are discussed in Chapter 436. 
 PATHOLOGIES ASSOCIATED WITH ABNORMAL BLOOD FLOW
PATHOLOGIES ASSOCIATED WITH ABNORMAL BLOOD FLOW
Kasabach-Merritt Syndrome
Kasabach-Merritt syndrome is a hemostatic complication of a vascular tumor. Diagnostic features included microangiopathic hemolytic anemia, thrombocytopenia, and hypofibrinogenemia. The definitive etiologic mechanism is still unknown. Using radioactively labeled platelets, several investigators have shown the evidence of platelet trapping.150-152 There is a hypothesis that the vascular endothelial cells of the lesion may have deficient barrier and anticoagulant function compared to normal blood vessels, thus causing excessive microvascular and intralesional thrombosis.103 These resulted in consumptive coagulopathy and also eventual resolution of the lesion.
Vascular Pathology
Vascular pathology such as cardiac valve abnormalities and sites of vascular stenosis can cause turbulent blood flow and high shear stress, which induces conformational change in circulating von Willebrand factor (vWF) to enhance binding affinity of vWF to unactivated platelets at the GP Ib sites.
Hemorrhagic Infarction
Bleeding symptoms or complications can occur with certain types of thrombosis, most commonly, hemorrhagic transformation in ischemic stroke. Cerebral hemorrhage within the infarcted tissue is a common complication after a focal ischemic injury. Hemorrhagic infarction is reported in 15% of children with arterial ischemic stroke,153 and two thirds of children with venous infract due to cerebral sinovenous thrombosis (CSVT).154 The phenomenon is now viewed as a consequence of the inflammatory reaction of an acute ischemic injury, causing disruption of the endothelial permeability barrier, as well as a consequence of an increase in arterial perfusion pressure and/or venous hypertension.155
SUMMARY
With regard to the hemostatic system, children are not “little adults,” and research has established that their coagulation and fibrinolytic systems are unique and dynamic. The hemostatic system in the young seems to protect them from thrombosis and bleeding tendencies, but they are not immune to either. The resultant morbidity and mortality can be severe. The clinical approach to treating neonates and children differs from adults because of the unique features of their hemostatic system. Unfortunately, randomized controlled studies for treating children are lacking, so at present, most recommendations are extrapolated from adult data, small case series, or observational studies.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


