Noncyanotic Congenital Heart Disease
INTRODUCTION
This chapter focuses on noncyanotic congenital heart disease with a basic description of the epidemiology, embryology, clinical manifestation, diagnostic testing, management, treatment, and prognosis of each lesion. The intent is to provide a framework for the major types of noncyanotic congenital heart disease, ranging from septal defects, left ventricular outflow tract (LVOT) obstruction disease, and right ventricular outflow tract obstruction disease. The chapter forms a guide for managing neonates with these common heart conditions.
SEPTAL DEFECTS
Atrial Septal Defect
The atrial septal defect (ASD) is one of the first forms of congenital heart disease that was corrected surgically. It represents a communication between the left and right atria and does not usually manifest in the neonatal period unless there is associated congenital heart disease, which usually makes it difficult to diagnose. There are 3 major types: secundum ASD, primum ASD, and sinus venosus ASD. The primum ASD is discussed as a part of the atrioventricular canal (AVC) defect (Figure 19-1).
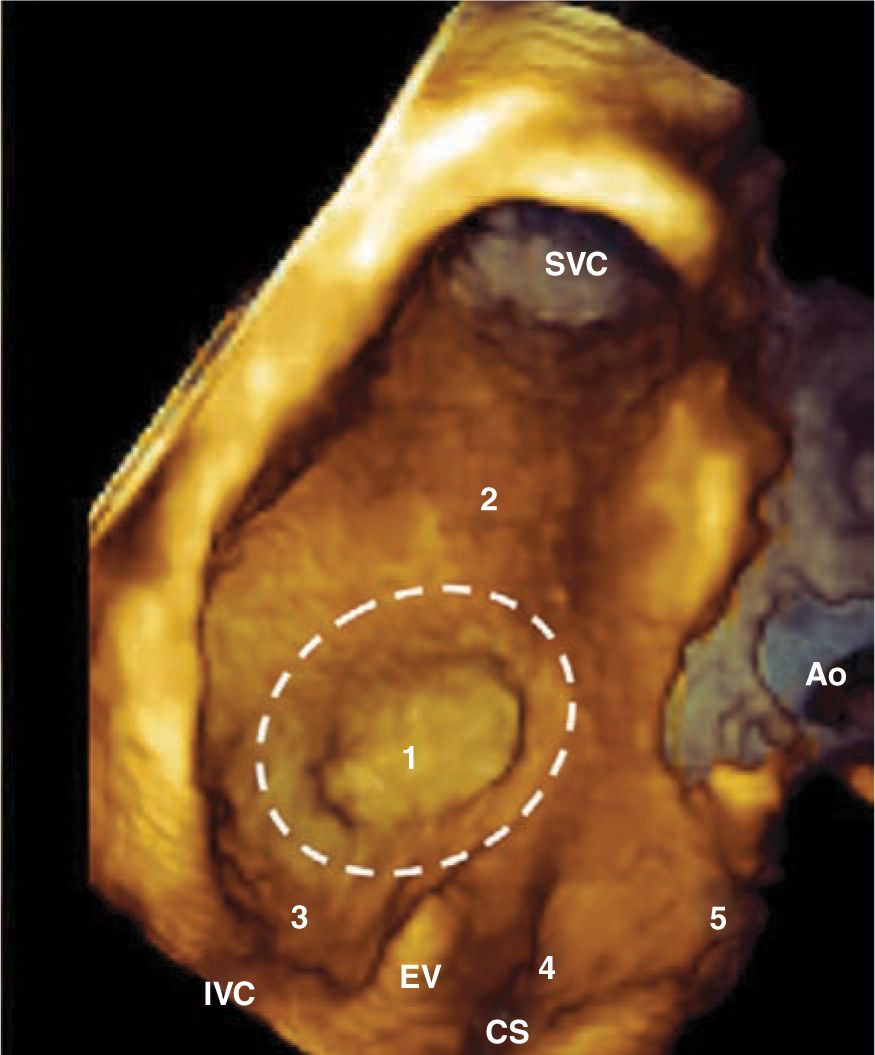
FIGURE 19-1 This 3-dimensional echocardiogram image depicts the interatrial septum from the perspective of the right atrial side. The superior vena cava (SVC) drains superiorly into the right atrium, while the inferior vena cava (IVC) drains from below. The fossa ovalis is central, and a secundum atrial septal defect (ASD) would lie in region 1. A superior or inferior sinus venosus ASD is located in regions 2 or 3, respectively. The primum ASD is located anteroinferiorly at region 5. The rare coronary sinus (CS) ASD, not discussed in this chapter, is located near the CS. The aorta (Ao), which is anteriorly located, is diagrammed as a point of reference. EV, Eustachian valve. (Reproduced with permission from Faletra et al.160)
Epidemiology
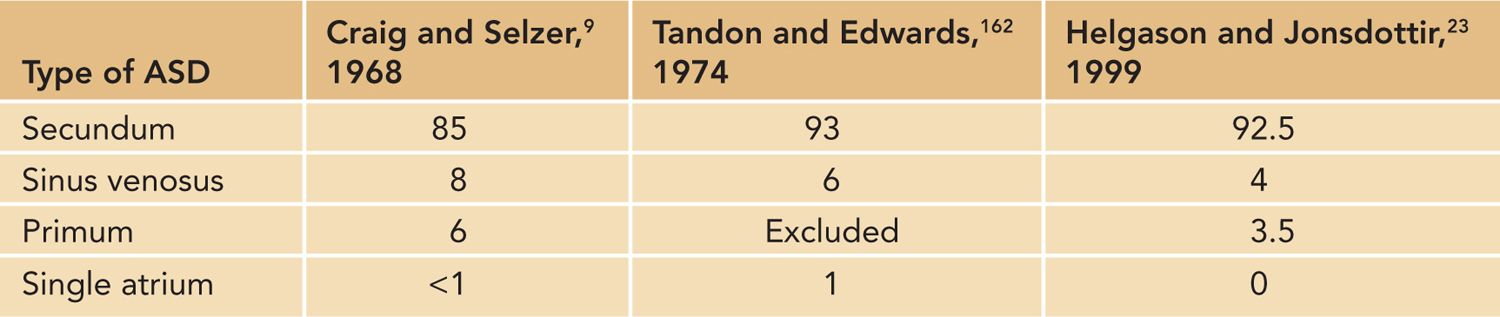
The ASD represents the second-most-common form of congenital heart disease behind the ventricular septal defect (VSD). The occurrence is between 3% and 4.1% of 1000 live births1,2 and 7% and 15% of all congenital heart disease cases.3,4 For defects that are larger and measure over 5 mm in diameter, there tends to be a female predominance.5,6 The secundum ASD is by far the most common type (Table 19-1). Nearly 1 of every neonates will have an atrial communication that is difficult to distinguish from a patent foramen ovale.7 Approximately 15% of trisomy 21 patients will have a secundum ASD, and 1% of ASD patients will have Holt-Oram syndrome.8 Of note, patients with Holt-Oram tend to have very large ASDs and can have a common atrium with no wall separating the atria.
Table 19-1 Common Forms of Atrial Septal Defect (ASD) (%)
Pathophysiology
Patients with ASDs that are hemodynamically significant tend to have problems later in life and do not present in the neonatal period. Specifically, there is a chronic left-to-right atrial shunt that enlarges the right atrium and right ventricle. This shunt increases blood flow to the pulmonary arteries, which chronically can change the pulmonary vascular bed, leading to pulmonary hypertension. Approximately 22% of ASD patients will develop pulmonary hypertension in adulthood if the ASD is left unrepaired, and 15% will have elevated pulmonary vascular resistance.9 Eisenmenger syndrome develops if a chronic left-to-right shunt lesion causes severe pulmonary vascular changes such that there is significant elevation in the pulmonary vascular resistance and a reversal of the shunt. Thus, blood would flow from the right atrium to the left atrium, resulting in arterial hypoxemia. Eisenmenger syndrome occurs 14% of the time in patients with an ASD.9 Again, if left untreated another complication is right heart failure from the chronic volume overload as well as atrial arrhythmias from right atrial distension.9 Fewer than 1% of infants with an isolated secundum ASD will develop significant symptoms that may lead to death.10
Infants can develop early right heart failure, which is presumably from a rapid drop in the pulmonary vascular resistance.11 The degree of shunting is related to the relative compliance of the left ventricle in comparison to the right ventricle. As the resistance in the pulmonary bed drops, the right ventricular “stiffness” decreases, leading to the left-to-right shunt. Thus, ASD should be considered in the differential diagnosis of any infant with congestive heart failure or failure to thrive.12
The secundum ASD represents a defect in the flap of the foramen ovale where it is incompetent, leading to left-right shunting. The sinus venosus ASD, however, is quite separate and high, where the right pulmonary veins and superior vena cava drain; frequently, there is no associated shunting across the foramen ovale. Thus, anomalous drainage of the right pulmonary veins into the superior vena cava is associated with the sinus venosus ASD.
Regardless of the type of ASD, most patients are asymptomatic in early childhood; however, congestive heart failure symptoms begin before the age of 6 years in 84% of patients and, as mentioned, symptoms can develop during infancy if there is a rapid drop in the pulmonary vascular resistance.9,11,13 The symptoms in the infant would include failure to thrive, tachypnea, feeding difficulties, and diaphoresis with feeds. Cyanosis would be unlikely because this defect does not produce Eisenmenger syndrome until adulthood. In the older patient, easy fatigability, syncope, and hemoptysis may also be present along with frequent pulmonary infections from the chronic left-to-right shunt. Unfortunately, 30% of children have only 1 or no typical physical sign from an ASD, which makes the diagnosis in the young child difficult.14
As a result of the extra volume of blood traversing the pulmonary valve, there is a relative pulmonary valve stenosis murmur. The systolic ejection-type murmur is present in 87% to 100% of the time.14,15 In addition, extra volume across the tricuspid valve may lead to diastolic murmur from relative tricuspid stenosis in some cases. As a result of this volume-loaded right ventricle, the pulmonary valve closes later than the aortic valve throughout the cardiac cycle, resulting in the “fixed” split in S2, which is present 57% of the time.14 Again, the neonate presents a challenge because in most patients the right ventricle is not volume loaded since the pulmonary vascular resistance remains relatively high and the right ventricle is “stiff.” Thus, the physical examination findings may not be present during infancy.12
A few etiologies have been discovered for the ASD. The only reported maternal exposure associated with ASD is maternal alcohol consumption, which appears to double the risk of ASD.16 Mutations in the fetus NKX2.5 gene have been identified in patients with many forms of congenital heart malformations, and ASD is common among them.17,18
Differential Diagnosis
Because the child with an ASD can have signs of congestive heart failure, other disease processes that cause congestive heart failure should be included in the differential diagnosis. Specifically, congenital heart disease that leads to a chronic left-to-right shunt should be considered, such as a VSD, patent ductus arteriosus, or an AVC defect. Infants with cardiomyopathy may also have the same symptoms noted for ASD. Finally, other noncardiac conditions that can cause congestive heart failure should also be considered, such as sepsis, severe anemia, or pulmonary infection.
Diagnostic Tests
Prior to the advent of echocardiography, the cardiac catheterization was used in patients with a clinical suspicion of ASD for diagnostic purposes, and repair was considered if the relative pulmonary blood was 50% greater than the flow across the aorta.3 The electrocardiogram (ECG) in the neonate and infant may not demonstrate the classic RSR′ pattern because this is a marker for volume load, and right ventricular hypertrophy is a common finding in normal neonates (Figure 19-2). Also, ECG findings are often missing in children with ASD.14
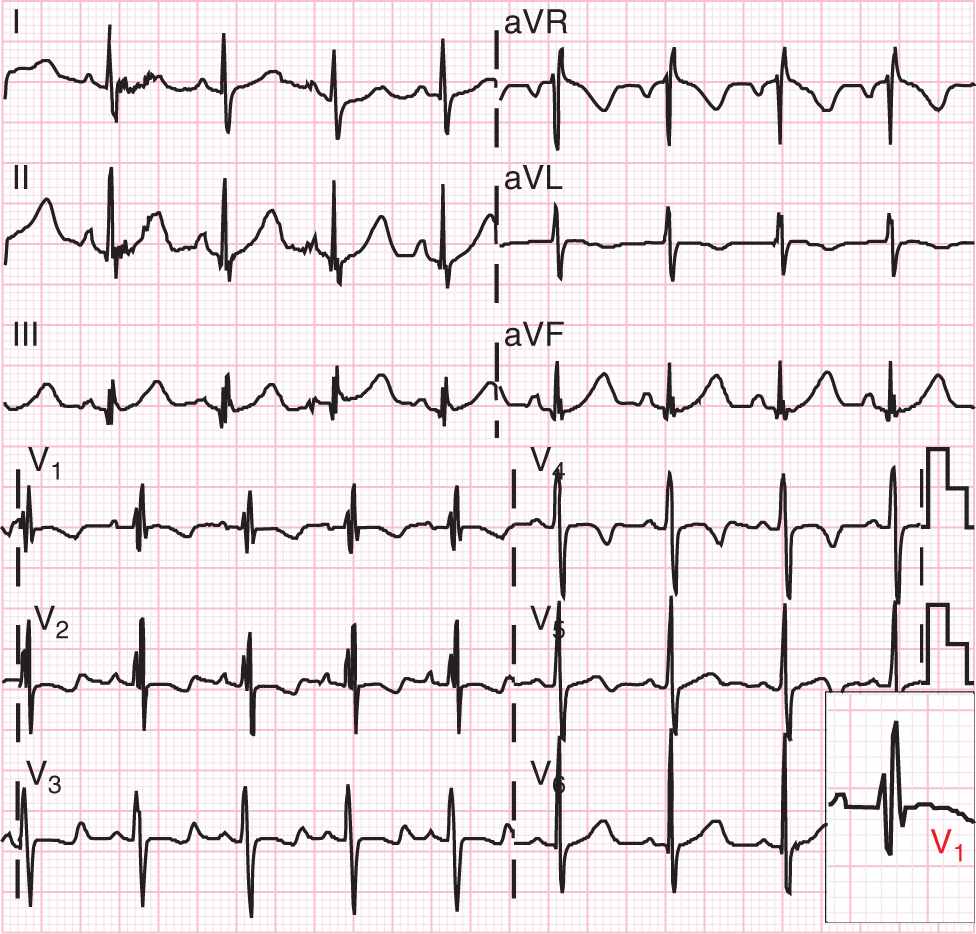
FIGURE 19-2 This electrocardiogram shows the classic RSR′ pattern in lead V1, which represents a volume load on the right ventricle. Note that the R′ is larger than the R in the bottom right inset.
Currently, the echocardiogram remains the gold standard for the diagnosis of all types of ASD. In addition to identifying the type of ASD, this technique is used to guide transcatheter closure and evaluate the surgical repair of an ASD.
Management
The decision to close a secundum ASD depends on the size of the defect, the degree of shunting, and the evidence of right ventricular enlargement. Traditionally, if the relative flow across the pulmonary artery is 50% more than the flow across the aorta because of the ASD, then an intervention is recommended.3 Surgical closure or modern-day device closure with interventional cardiology of secundum ASDs is generally low risk and curative. The sinus venosus ASD is surgically repaired because there is no intervention available for this defect at this time.
In general, the closure of an ASD should be done in early childhood; however, as mentioned, there are cases where an infant would have evidence of congestive heart failure and secondary failure to thrive, which would warrant an earlier intervention.19 Infants can also safely undergo ASD device closure in the catheterization laboratory, which would avoid a sternotomy and cardiopulmonary bypass.20
Spontaneous closure occurs in 26% of patients before the age of 2 years, and in general an ASD is unlikely to close after this age, which supports the idea of repair in early childhood.21 Ninety-eight percent of ASDs less than 3 mm in diameter in neonates close by 18 months of age, and defects greater than 3 mm take longer to close.6,22 Thus, size of the defect at initial presentation has an impact on the spontaneous closure rate. The ASDs in premature infants take longer to close; however, an associated patent ductus arteriosus tends to close the ASD sooner, presumably because of elevated left atrial pressure, which causes the septum primum flap to abut against the septum, resulting in closure of the defect.6
Outcome and Follow-up
Death is rare in patients with unrepaired ASD; however, atrial arrhythmias in adult patients with chronic atrial distension from the left-to-right shunt is much more common.3 Cerebrovascular accidents are even rarer than mortality but still may occur.3 Most patients are not symptomatic until the third decade of life; however, intervention is recommended in early childhood.3,9
With the advent of echocardiography and color Doppler, ASDs are recognized more readily at an earlier age. Over 70% to 80% of all secundum ASDs close spontaneously by 18 months of age.1,5 A defect less than 3 mm in diameter has a nearly 100% spontaneous closure rate, while a defect greater than 8 mm is unlikely to close,8,23 confirmed by later studies.24–26
Ventricular Septal Defect
The VSD represents a communication between the right and left ventricles and varies significantly in presentation. For the most part, the neonate may not have symptoms from a VSD even when it is large because flow across it relies on a drop in the pulmonary vascular resistance. The defect is classified by its location within the ventricular wall, the size, and the number of defects. Essentially, there are 3 major types: perimembranous, doubly committed subarterial (supracristal), inlet, and muscular (Figure 19-3). The perimembranous VSD lies superior and anterior within the region of the membranous septum, which is bordered by the tricuspid valve and aortic valve. The supracristal VSD is also anterior but represents a defect below the pulmonary and aortic valves. Both of these defects are outlet VSDs. The inlet VSD is a defect that is near the atrioventricular valves and located quite posteriorly. This defect is most commonly discovered with a complete atrioventricular canal (CAVC) defect, which is discussed further in this chapter. The muscular VSD represents a defect in the trabecular end of the ventricular septum and is bordered completely by muscle. Any combination of these defects may coexist; thus, the clinician should always be suspicious of multiple defects.
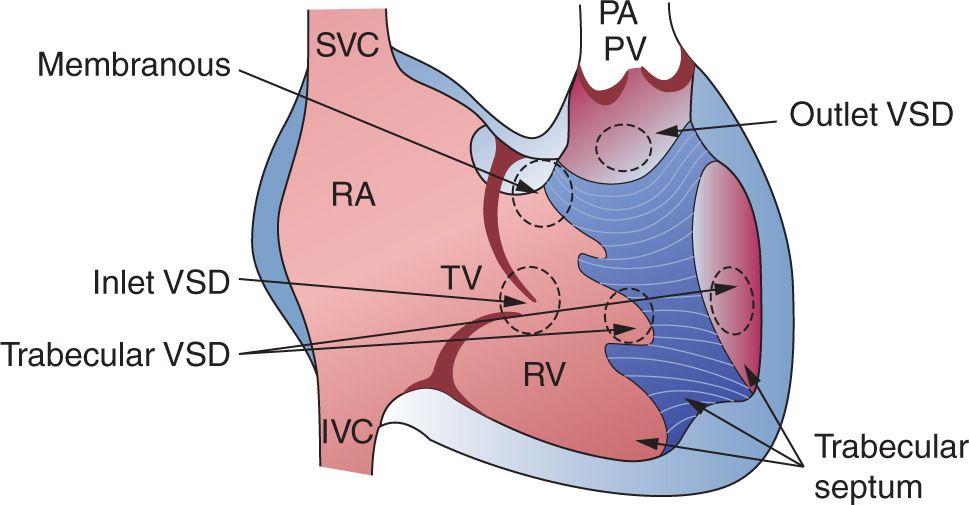
FIGURE 19-3 This diagram of the right ventricular aspect of the interventricular septum demonstrates the different types of ventricular septal defects (VSDs). Note that the inlet VSD is near the atrioventricular valves, and the outlet VSD tends to be near the outflow tract. IVC, inferior vena cava; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava; TV, tricuspid valve. (Reproduced with permission from Axt-Fliedner et al.40)
Epidemiology
The VSDs occur approximately in 0.95 of 1000 live births and represent the most common form of congenital heart disease other than bicuspid aortic valve. The majority of defects are perimembranous, ranging between 62% and 83%, followed by muscular defects, which vary between 9% and 12%.27–30 Supracristal defects are much less common and occur between 2% and 3% of the time.27,29 Muscular defects are less likely to be associated with a syndrome or a karyotype anomaly.1
Pathophysiology
The primary issue with the VSD is related to the degree of shunting from the left to the right ventricle. Multiple variables influence the degree of shunting and resultant symptoms from this type of defect. Specifically, the size and location of the defect are important in that small defects tend to protect against congestive heart failure. However, multiple small defects, known as a “Swiss cheese” VSD, may cause significant heart failure because of the collective size of all of the defects. Swiss cheese–type defects account for 0.5% of VSDs.28 The other variable of importance is the relative resistance of the systemic and pulmonary vascular bed. In the neonatal period, the pulmonary vascular resistance is high; therefore, there is minimal shunting. As the pulmonary vascular resistance falls dramatically in the first few weeks of life, more left-to-right systolic shunting occurs across the defect. Because the relative pressures in diastole are similar between the ventricles, there is minimal diastolic shunting. Blood that shunts in systole enters directly into the main pulmonary artery; therefore, there is no nidus for right ventricular enlargement. The main pulmonary artery, left atrium, and left ventricle dilate with VSD physiology but not the right heart structures.
As the pulmonary vascular resistance drops, infants develop symptoms, which include tachypnea, feeding intolerance, failure to thrive, and diaphoresis.27,31 However, 50% of infants may have no symptoms despite having a large defect. If the pulmonary vascular resistance fails to drop significantly, then the shunting and congestive heart failure symptoms would be minimal. Of note, premature infants tend to go into congestive heart failure much quicker than term infants; however, the proportion of patients in either group with these symptoms is the same.31 The pulmonary vascular resistance drops with age, as expected in the setting of a VSD, but the resistance is generally higher for larger VSDs.31
The VSD murmur is usually not heard in the first few days of life unless it is a small defect. In fact, the murmur typically occurs within the first week to month of life in most cases.31 Classically, the VSD’s murmur is holosystolic in that it blends with the first heart sound, which generally cannot be auscultated. In addition, there may be excessive flow across the pulmonary and mitral valves, which would lead to a relative stenosis murmur in systole and diastole, respectively. In the case of a large VSD, the murmur may be difficult to hear.
Spontaneous closure of the VSD has been noted for the perimembranous and muscular VSD through various mechanisms. Tricuspid valve tissue may adhere to the perimembranous defect, resulting in occlusion of the defect over time.32,33 The aortic valve sinus of Valsalva may also occlude the defect over time; however, this may be associated with herniation or rupture of the sinus into the right ventricle or aortic insufficiency. The margins of the muscular defect become relatively small as the child grows and the interventricular septum thickens, which is believed to be the mechanism of spontaneous closure.
Many associations have been noted with the outlet VSDs. Specifically, there can be a subaortic membrane or ridge associated with the defect that causes stenosis.28,34 As noted, aortic insufficiency is associated with perimembranous defects but is also associated with supracristal defects because of the lack of tissue supporting the subvalvar apparatus.28,35 However, subacute bacterial endocarditis associated with having a high-velocity jet and endothelial damage has also been noted to occur in 6% of all VSDs and can lead to aortic insufficiency secondary to direct damage; this will only exaggerate the congestive heart failure symptoms.36 Subacute bacterial endocarditis and aortic insufficiency tend to be manifestations of a VSD later in life; congestive heart failure begins in infancy. An additional association is double-chamber right ventricle, for which a callous formation develops on the opposing wall of the right ventricle, which causes subpulmonary valve obstruction. Any outlet VSD can have an association with a coarctation of the aorta, which is discussed further in this chapter. VSD has an association with other forms of more complex cyanotic congenital heart disease; discussion of this is beyond the scope of this chapter.
The associated noncardiac conditions include trisomy 21, the VACTERL (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities) association of congenital defects, cleft palate, bronchopulmonary dysplasia, chondrodysplasia, Klippel-Feil syndrome plus omphalocele, and polysplenia syndrome.37
Differential Diagnosis
Given that the VSD presents with signs of congestive heart failure in the infant after decreases in pulmonary vascular resistance, any other disease process that causes congestive heart failure must be ruled out. As with the ASD differential diagnosis, the left-to-right shunt lesions must be considered, such as AVC, patent ductus arteriosus, and aortopulmonary window. Also, as the pulmonary vascular resistance drops, infants with anomalous origin of the coronary artery from the pulmonary artery would present as well because of coronary ischemia and steal. These patients would present with heart failure symptoms as well. In addition, lesions that cause high-output failure can mimic symptoms of a VSD. These include and are not limited to sepsis, severe anemia, and hepatic and cerebral arteriovenous malformations.
Diagnostic Tests
Ascultatory findings may not be evident on large defects because a VSD murmur may not be present. The ECG will demonstrate left atrial enlargement and left ventricular hypertrophy in the setting of large defects; chest x-ray will show evidence of cardiomegaly with increased pulmonary vascular markings. These findings, however, are nonspecific, and the diagnosis is usually made by echocardiography.38 In fact, detection and closure of a VSD can be determined by fetal echocardiography as well.39,40 In the past, these defects were confirmed by cardiac catheterization, which is no longer necessary.
Management
Large VSDs need to be repaired during infancy, and the elevated pulmonary vascular resistance as a result of the chronic shunt should improve postoperatively.27 There is no difference in outcome for large VSD closure in children less than 4 kg vs greater than 4 kg; thus, it is recommended to intervene early to prevent recurrent respiratory infection, failure to thrive, and feeding difficulties.37
In the past, a pulmonary artery band (PAB) was positioned to limit pulmonary blood flow, and the VSD was closed as a separate procedure to minimize morbidity and mortality; however, VSDs are now closed in 1 step with primary closure regardless of the type. The indications for surgery include left heart failure, failure to thrive, feeding problems, and elevated pulmonary artery pressure. VSDs are usually closed through the right atrium41,42 as opposed to a ventriculotomy, which can create a scar on the myocardium. For defects closer to the semilunar valves, repair can also occur through the pulmonary valve.43 Multiple muscular VSDs can be closed primarily instead of with a PAB; however, if the septum is Swiss cheese, a PAB may be necessary because a patch across all of the defects may be difficult and may result in ventricular dysfunction.44 After PAB placement, the defects may close spontaneously. Perventricular device closure of muscular VSDs in the cardiac catheterization lab may be an option in some select patients45,46; however, closure of perimembranous VSDs was not available when this chapter was written. This option is attractive to avoid a sternotomy, PAB, or cardiopulmonary bypass.
Outcome and Follow-up
The natural history of a VSD is for spontaneous closure, reduction in size, or development of pulmonary vascular disease. Death in infancy is most probably related to congestive heart failure.47 The overall mortality is approximately 3% in all patients.36 In general, muscular defects tend to have less congestive heart failure, higher closure rate, and less need for operative intervention compared to outlet defects.48
The natural closure rate for all VSDs is 22% to 36%.31,33,49 The vast majority of defects that close do so before the eighth year of life and tend to be less than 6 mm in diameter.49 Approximately 15% to 29% of perimembranous defects close compared to 57% to 65% of muscular defects.1,28,50 Muscular defects tend to close during the first 6 to 12 months of life.51 The most common complications after cardiac surgery include a residual VSD, right bundle branch block, complete heart block, death, persistent pulmonary hypertension, cerebrovascular accident, or tricuspid valve regurgitation, which may be related to perimembranous patch closure. However, in a recent article, the surgical results for isolated VSD were noted to be good, with 0% reoperation for residual defect, 0.5% mortality, 99.5% of patients asymptomatic, and none had more than mild new-onset tricuspid valve regurgitation.30
Atrioventricular Septal Defect
An atrioventricular septal, or canal, defect represents a malformation of the AVC whereby the crux of the heart does not develop, which potentially renders a primum ASD and inlet VSD. Thus, there is a spectrum of disease ranging from an isolated primum ASD with a cleft mitral valve to a CAVC defect with a large inlet VSD and primum ASD (Figure 19-4). As a result of these communications, patients can develop a chronic left-to-right shunt and atrioventricular valve regurgitation. As with other ASDs, a patient with an isolated primum ASD and cleft mitral valve does not usually present with heart failure in infancy; however, a CAVC can cause significant congestive heart failure in the first few months of life.
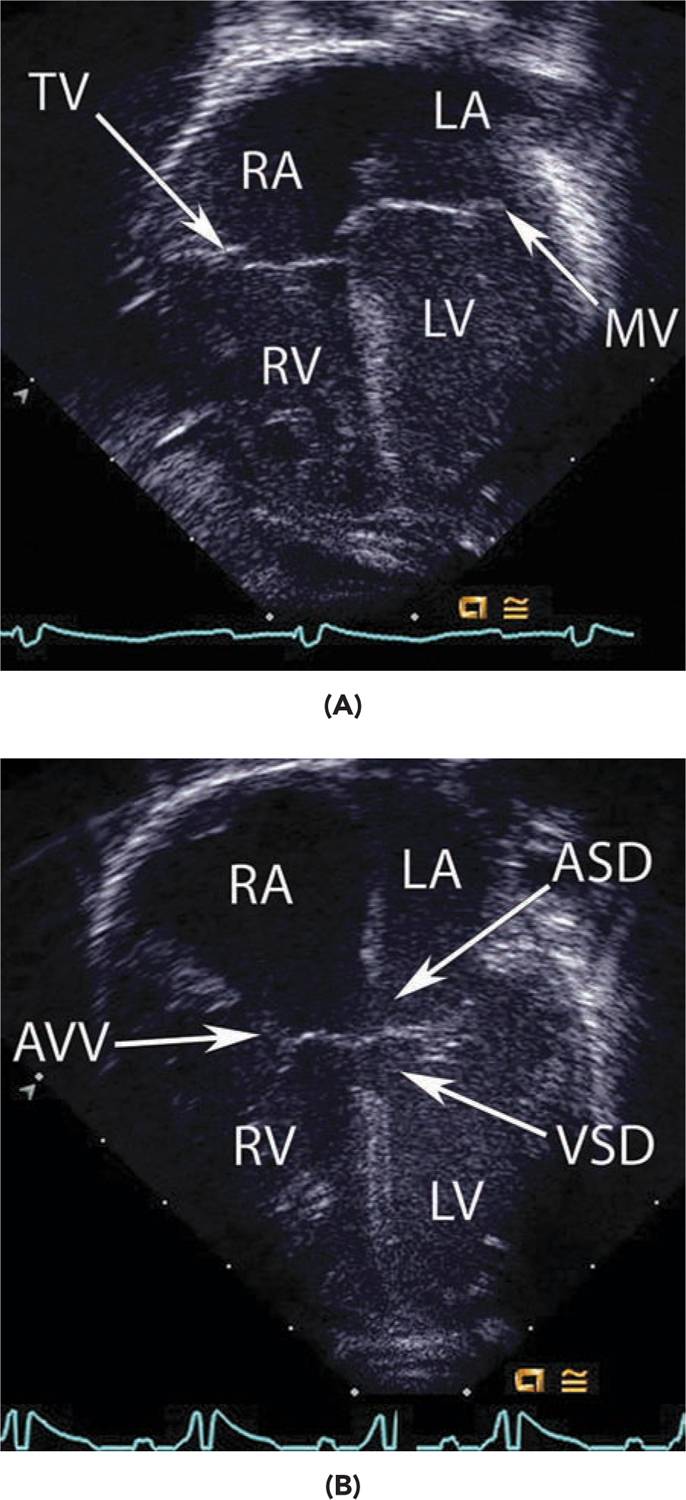
FIGURE 19-4 A, A normal 4-chamber view in diastole obtained by echocardiography. Note the atrial wall separating the right (RA) and left atrium (LA). The tricuspid valve (TV) is slightly lower and closer to the apex compared to the mitral valve (MV). LV, left ventricle; RV, right ventricle. B, This image shows a patient with a complete atrioventricular canal defect. There is a common atrioventricular valve that is not offset like the normal heart. A large atrial septal defect (ASD) and inlet ventricular septal defect (VSD) are also noted.
Epidemiology
The diagnosis of AVC defect often occurs with trisomy 21. In fact, about 50% of patients with trisomy 21 and congenital heart disease have this defect. Specifically, 36% of patients with trisomy 21 have AVC defects.52
The AVC defect represents the most common form of heart disease in trisomy 21,53 followed by VSD, ASD, and then tetralogy of Fallot. Importantly, about 40% to 50% of patients with Down syndrome have congenital heart disease in general.52,54 For trisomy 21, a CAVC defect is more common than an isolated primum ASD with cleft mitral valve, which only occurs in 3% to 25% of patients with isolated primum ASD and cleft mitral valve series.13,52,55 A CAVC defect can also occur in the setting of tetralogy of Fallot, which is associated with a genetic abnormality about 88% of the time, 67.2% of which is trisomy 21.56
Pathophysiology
As with the VSD and ASD, the shunt for a CAVC or isolated primum ASD is related to a drop in pulmonary vascular resistance. Thus, neonates may not have symptoms unless there are associated abnormalities. As the pulmonary vascular resistance falls, the infant can have symptoms of tachypnea, diaphoresis, and feeding difficulties, which can lead to failure to thrive. Of course, the VSD physiology of the AVC has a higher likelihood of causing symptoms than an isolated primum ASD with mitral valve cleft because the pulmonary arterial bed is under a pressure load and not just a volume load.
In addition to the left-to-right shunt, patients may have significant left atrioventricular, mitral valve, regurgitation, which elevates the left atrial pressure and can lead to worsening pulmonary venous congestion and respiratory symptoms. As with any of the left-to-right shunts, patients are at risk of recurrent respiratory infections if left untreated. Given that a large portion of patients with AVC have trisomy 21, it is important to note that children with trisomy 21 can have persistently elevated pulmonary vascular resistance in the first year of life compared to the normal patient population, which would make them potentially asymptomatic with no significant shunt.57 However, children with trisomy 21 have a higher risk of fixed pulmonary vascular disease compared to children without trisomy 21, making the need for repair even greater.57
Several important associated anomalies must be accounted for when treating neonates with AVC. If the canal is unbalanced, the atrioventricular valve favors 1 ventricle over the other, making the other somewhat hypoplastic. In some cases, the ventricles are so unbalanced that the hypoplastic chamber is inadequate to serve the needs of the systemic or pulmonary circulation. Most of the time, this is an unbalanced left ventricular AVC, which renders hypoplastic left heart syndrome. Coarctation of the aorta, atrioventricular valve regurgitation, LVOT obstruction, and tetralogy of Fallot (discussed previously) are other conditions associated with AVC defects. Rarer anomalies would include heterotaxy with either right or left atrial isomerism, double-orifice mitral valve,58–61 and cor triatriatum, a membrane that obstructs flow into the left ventricle.62,63
Differential Diagnosis
The AVC defect usually presents with symptoms of a chronic left-to-right shunt just like the ASD or VSD; however, the patients may present sooner given the multiple levels of shunting and the association with other congenital heart disease noted previously. Thus, these patients can present with congestive heart failure, and other diseases that present in this manner must be excluded. Namely, noncardiac conditions would include sepsis, severe anemia, and hepatic and cerebral arteriovenous malformations, and an additional cardiac condition would be a patent ductus arteriosus or aortopulmonary window.
Diagnostic Tests
The ECG in patients with AVC defect have an unusual counterclockwise direction of the frontal electrical conduction vector, resulting in the classic northwestern QRS axis52 (Figure 19-5). Thus, a patient with this classic ECG finding along with the stigmata of trisomy 21 has an AVC defect until proven otherwise. Nonetheless, the echocardiogram can help determine the diagnosis more specifically. In addition, the associated lesions, such as balancing of the ventricles, valve regurgitation, left or ventricular outflow tract obstruction, and coarctation of the aorta, can be entertained. The typical appearance of the AVC is readily recognized from the 4-chamber view (Figure 19-4). The cardiac catheterization is reserved for those cases where the pulmonary vascular resistance is called into question if a child does not receive a repair early in infancy.
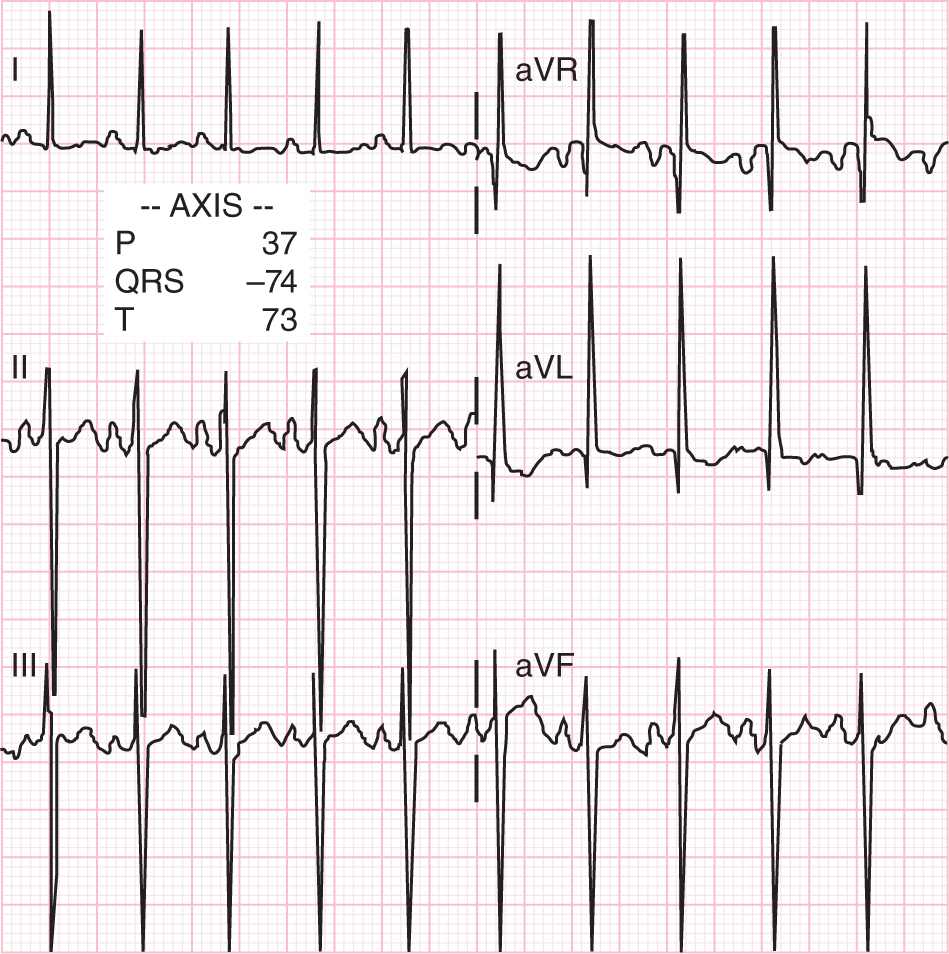
FIGURE 19-5 Classic electrocardiogram for a patient with complete atrioventricular canal. Notice that the QRS axis derived from these limb leads is −78°.
Management
Initial management in the past was PAB placement in infancy because of the poor results in infancy from CAVC defect repair.64 Now, with improved techniques, surgery occurs predominantly in infancy without the need for a PAB. In the setting of a CAVC defect, there is no reason to wait beyond the 3-month age group to repair if there is a large VSD, which will only cause pulmonary vascular disease, especially in the large subgroup of trisomy 21 patients with pulmonary resistance issues from the start.57 A primum ASD with a cleft mitral valve can be corrected later in life, and the timing of surgery is dependent on the institution’s practice. At the time of repair, the primum ASD and inlet VSD are repaired along with an associated lesion, with patching of the defects and repair of a cleft in the mitral valve. In the past, the CAVC defect was repaired late in infancy; however, improved morbidity and mortality associated with earlier repair supports the idea of not delaying surgical intervention.65
Outcome and Follow-up
Without surgery, the survival for CAVC defect is 54% at 6 months of age and 35% at 12 months of age, 15% reach 24 months, and 4% reach 5 years of age66; thus, surgery is recommended for the children at risk for florid heart failure as the pulmonary vascular resistance decreases and there are multiple shunts. Heart failure in combination and severe respiratory infections are the modes of death among patients with trisomy 21.52 However, there is no difference in outcomes comparing the patients with trisomy 21 with the nonsyndromic patients for AVC defect, ASD, or VSD lesions, which eventually lead to more children with trisomy 21 having congenital heart disease surgery.53,67 The operative mortality has decreased significantly,68–70 from 25% before 1976 to less than 4% after 1987. Complications after repair include death, 1.6%; postpericardiotomy syndrome, 13%; atrial arrhythmia, 3%; transient heart block, 1.6% to 3.5%; sternal wound infection, 0.5%; and left hemiparesis, 0.5%.55,70 The 10-year survival is 98% in patients with AVC repair.55 Some patients will require reintervention for mitral valve regurgitation (4%–7%)70,71 or LVOT obstruction.
LEFT VENTRICULAR OUTFLOW TRACT ABNORMALITIES
Aortic Stenosis
The aortic valve in patients with aortic stenosis can have a variable morphology, appearing dysplastic, thickened, bicuspid, or hypoplastic. As a result of this aortic valve disease, neonates tend to present with variable degrees of obstruction; however, it is rare for infants to have significant aortic insufficiency without intervention. The presentation of aortic stenosis can be variable and depends on the degree of obstruction. In the most severe examples, the neonate is ductal dependent for systemic blood flow because of inadequate output from the left ventricle, and there is retrograde flow from the ductus arteriosus and aortic arch that feeds the brachiocephalic vessels and descending aorta. This clinical presentation is an example of critical aortic stenosis; however, much milder forms of aortic stenosis render the neonate asymptomatic.
Epidemiology
Aortic stenosis represents approximately 6% of all congenital heart disease and occurs in 6 of 10,000 live births.72 Twenty-five percent of cases will also have a fibrous or muscular subaortic stenosis associated with the aortic stenosis. Rarely, this condition can be associated with pulmonary stenosis, especially in the setting of congenital rubella syndrome.73 Genetic syndromes are also known to be associated with aortic stenosis and include Turner, Goltz, Costello, and Williams syndrome. If these genetic syndromes are suspected, an evaluation for aortic stenosis and other congenital heart disease should be initiated.
Pathophysiology
As discussed, the presentation of aortic stenosis will vary depending on the degree of obstruction, which can be mild to severe. The aortic valve leaflets typically are bicuspid and thickened with a decrease in cross-sectional area and can have associated supravalvar or subvalvar narrowing. Because of the obstruction to the aortic valve, the left ventricle can be hypertrophied in accordance with the degree of stenosis. In some cases with severe obstruction, the left ventricle begins to fail, resulting in a dilated ventricle with diminished systolic function. As a result of the high afterload, there can be endocardial fibroelastosis, which can impair both systolic and diastolic function of the left ventricle. With impaired diastolic function, the left ventricle fails to relax, resulting in high end-diastolic pressure and thus high left atrial and pulmonary arterial pressure from the back pressure.
The other common associated lesions include patent ductus arteriosus, coarctation of the aorta, VSD, ASD, mitral stenosis, and Shone’s complex.73,74 Shone’s complex is a heart condition with multiple left heart obstruction lesions that traditionally include parachute mitral valve (single papillary muscle), supravalvar mitral ring, congenital mitral stenosis, subaortic stenosis (and valvar aortic stenosis), and coarctation of the aorta. Eight percent of patients with aortic stenosis have Shone’s complex.75 Often with Shone’s complex, the left ventricle is deemed inadequate to handle the work of the systemic circulation, and the neonate must undergo a Norwood procedure for hypoplastic left heart syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


