Non-Hodgkin Lymphoma
Mitchell S. Cairo and Thomas G. Gross
 EPIDEMIOLOGY
EPIDEMIOLOGY
Lymphoma represents the third most common pediatric cancer in children between the ages of 0 to 19 years of age.1 Sixty percent of all childhood and adolescent lymphomas are classified as non-Hodgkin lymphoma (NHL). NHL represents approximately 8% to 10% of all malignancies that occur in children between ages 5 to 19 years. Approximately 750 to 800 cases of NHL are diagnosed annually in children and adolescents younger than age 20 years in the United States.  Risk factors for the development of NHL in children and adolescents are largely unknown, except that children with either inherited and/or acquired immunodeficiencies have a significantly increased risk for developing NHL.
Risk factors for the development of NHL in children and adolescents are largely unknown, except that children with either inherited and/or acquired immunodeficiencies have a significantly increased risk for developing NHL.
 PATHOLOGY
PATHOLOGY
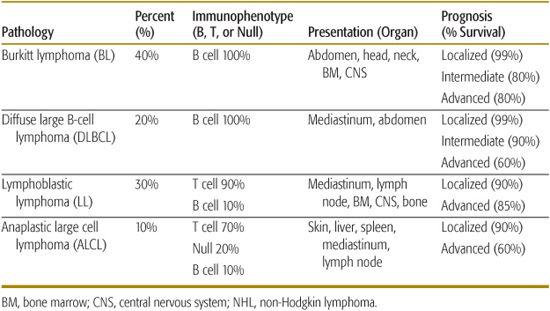
As opposed to non-Hodgkin lymphoma (NHL) seen in adults, over 95% of childhood and adolescent NHL has been classified to be either an intermediate or aggressive pathological subtype by the World Health Organization (WHO) classification.5 The WHO classification, which is the most recent classification for NHL, incorporates morphological, immunophenotypic, and molecular characteristics to subclassify each type of lymphoma. There are four major pathological subtypes of childhood NHL and the distribution of these four histological subtypes include Burkitt lymphoma (BL) (40%), diffuse large B-cell lymphoma (DLBCL) (20%), lymphoblastic lymphoma (LL) (30%) and anaplastic large-cell lymphoma (ALCL) (10%) (Fig. 451-1).2,3,6
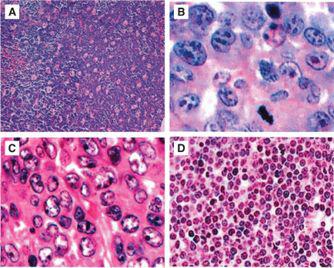
FIGURE 451-1. Hematoxylin and eosin staining of morphology of the 4 most common subgroups of childhood and adolescent non-Hodgkin lymphoma (NHL): (A) Burkitt lymphoma (BL)—high power, (B) diffuse large B-cell lymphoma (DLBCL)—high power, (C) precursor T-lymphoblastic lymphoma—high power, (D) anaplastic large cell lymphoma (ALCL)—high power.
 CLINICAL PRESENTATION, DIAGNOSTIC EVALUATION, AND STAGING
CLINICAL PRESENTATION, DIAGNOSTIC EVALUATION, AND STAGING 
Less than 10% of childhood and adolescent NHL presents in peripheral nodal tissue; the majority presents either in extranodal lymphoid tissue, including the thymus or Peyer patches within the gastrointestinal tract, or manifests in nonlymphatic tissue such as bone marrow, spinal fluid, skin, bone, gonads, or within the central nervous system (CNS). The most common sites of presentation include head and neck (30%), abdomen (30%), and mediastinal and/or hilar lymph nodes (30%). Each pathological subtype has distinct primary organ sites of presentation as outlined in Table 451-1. In general, children with either Burkitt lymphoma (BL) or lymphoblastic lymphoma (LL) have a higher prevalence for involvement of the bone marrow and central nervous system (CNS). Children with either diffuse large B-cell lymphoma (DLBCL) or LL have a higher prevalence for mediastinal and/or hilar nodal involvement (Table 451-1). Anaplastic large cell lymphoma (ALCL) has the highest prevalence for involvement of the skin. There are two common sub-forms of BL in children, the sporadic form which occurs more commonly in North America and Europe, which is only occasionally associated with Epstein-Barr virus (EBV) and the endemic form which occurs in children in equatorial Africa, which is uniformly associated with EBV. The endemic form in equatorial Africa more commonly presents in children with head and neck and particularly jaw, involvement. Comparatively, the sporadic form occurs with abdominal and much less commonly with head and neck, involvement.2
Table 451-1. Childhood and Adolescent NHL: Pathology, Distribution, Immunophenotype, Presentation, and Prognosis
Diagnostic work-up of children or adolescents suspected of having NHL includes a variety of studies and tests to determine diagnosis and stage of disease. Because hematologic dissemination is common in all types of pediatric NHL, a diagnostic lumbar puncture, bone marrow aspirate, and biopsy are required at diagnosis (eTable 451.2  ).
).
The Ann Arbor staging system for lymphoma is less useful in predicting outcome due to the propensity for extranodal involvement, noncontinuous spread of disease, and a higher propensity for metastatic spread to the bone marrow and central nervous system (CNS) in children with NHL. Therefore, the St. Jude’s staging classification is used for childhood NHL10,11 Limited disease includes stage 1 and 2. The more advanced disease includes stage 3 and stage 4. Approximately 70% of children and adolescents with NHL have stage 3 or 4 at the time of diagnosis (eTable 451.1  ).
).
 GENERAL PRINCIPLES OF THERAPY
GENERAL PRINCIPLES OF THERAPY
The primary therapeutic modality for all pathological subtypes of childhood and adolescent non-Hodgkin lymphoma (NHL) is multiagent chemotherapy. Current studies are investigating the potential role of targeted immunotherapy or potentially, even small molecule inhibitors for specific targeted therapy. The role of surgery is quite limited in the treatment of childhood and adolescent NHL, however it is critically important in establishing the diagnosis and to stage the disease. Currently, radiotherapy is not used in the treatment of pediatric NHL except when required for emergency treatment in the management of severe venocaval disease, large mediastinal masses precipitating acute respiratory distress, and acute CNS or peripheral nerve compression.
Another common complication, particularly in patients with BL or LL, is the development of tumor lysis syndrome (TLS).12 Though TLS usually occurs following initiation of therapy, it may develop before therapy is started. Rapid lysis of tumor cells may induce severe metabolic derangements, including hyperkalemia, hyper-phosphatemia, hypocalcemia, and hyperuricemia, This may lead to acute complications including renal failure, cardiac arrhythmias and dysfunction, and/or severe neurological complications.12 The mainstay of prophylaxis and/or treatment of TLS include hydration, frequent monitoring of electrolytes, and diuresis. Prevention and treatment of hyperuricemia requires either allopurinol, a competitive inhibitor of xanthine oxidase, which can prevent new uric acid formation, or rasburicase, a recombinant urate oxidate, which metabolizes uric acid to allantoin, significantly more soluble than uric acid in an acidic urine  .12,13
.12,13
 SPECIFIC TREATMENT FOR DIFFUSE LARGE B-CELL LYMPHOMA AND BURKITT LYMPHOMA
SPECIFIC TREATMENT FOR DIFFUSE LARGE B-CELL LYMPHOMA AND BURKITT LYMPHOMA
Treatment strategy for diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL) in childhood and adolescence is essentially the same. A recently developed French, American, and British (FAB) classification has improved the staging of childhood and adolescent B-cell non-Hodgkin lymphoma (NHL). It was recently applied in a large international study of children with mature B-cell lymphoma (LMB-FAB/96).14,15 With current therapeutic approaches, the prognosis of childhood and adolescent B-cell NHL is excellent. Patients with limited stage disease (Group A) show approximately 99% overall survival and require only 3 weeks of therapy. Those with intermediate risk disease (Group B) have a 90% survival with approximately 12 weeks of therapy. Patients with advanced disease, that is, bone marrow or CNS involvement (Group C), have an estimated 80% overall survival with 4 to 6 months of therapy (Table 451-1).14,15,23,29,31
It is very difficult to control recurrent or refractory B-cell NHL even with very aggressive chemotherapy and/or hematopoietic stem cell transplantation (HSCT). Survival rate for patients with recurrent or refractory disease is less than 25%.14,15,20,21
New directions in the diagnosis and pathogenesis include an increased understanding of the biology of childhood and adolescent B-cell NHL with the identification of new surface targets,34,35 recognizing the specific cytogenetic aberrations associated with childhood and adolescent B-cell NHL,36 specific pathological subtypes of diffuse large B-cell lymphoma (DLBCL),37 and the molecular subtypes of DLBCL and Burkitt lymphoma (BL) by oligonucleotide microarray molecular profiling.38,39
ANAPLASTIC LARGE-CELL LYMPHOMA
Adolescent anaplastic large-cell lymphoma (ALCL) was originally identified in 1985 as a unique subtype of non-Hodgkin lymphoma (NHL) represented by large pleomorphic blasts with a constant expression of a surface protein CD30 (Ki-1).45 Diagnosis of ALCL can be difficult due to waxing and waning symptoms, and can present as Hodgkin lymphoma and diffuse large B-cell lymphoma (DLBCL). The World Health Organization (WHO) classification of lymphomas recognizes two subtypes of ALCL, a primary cutaneous subtype and a systemic nodal and extranodal subtype.5,47 Primary cutaneous and ALK-negative ALCL have a very good prognosis requiring little therapy beyond resection. However, they are very rare in children and adolescents. Children and adolescents with localized systemic ALCL have an estimated 90% overall survival compared to patients with advanced disease, who have an estimated 60% to 70% overall survival (Table 451-1).50-57 Patients with advanced stage ALCL with skin involvement and mediastinal and/or visceral involvement including spleen, liver, or lung have a poor prognosis compared to other patients with advanced disease stage ALCL.58
For patients that have refractory or progressive disease, the prognosis is intermediate with overall survival ranging between 30% and 70%, but requires aggressive chemotherapy and usually bone marrow transplant (BMT). New approaches in the diagnosis, pathogenesis, and treatment of childhood and adolescent systemic ALCL include identifying specific protein targets that are distinct in ALCL63 and in particular potential downstream targets within the ALK downstream network. 
LYMPHOBLASTIC LYMPHOMA (LL)
Lymphoblastic lymphomas (LLs) can be of either T-cell or B-cell lineage, but more than 75% have T-cell markers. The patient with LL can represent a medical emergency. These tumors can double in size in a matter of days, Therefore, tumor lysis syndrome (TLS) is common. Also, about 75% of patients with LL have an anterior mediastinal mass. Depending on the age of the patient and compressibility of the trachea and bronchi, manifestations may be predominantly respiratory, such as dyspnea, wheezing, stridor, or dysphagia, rather than circulatory, that is, swelling of the head and neck. Pleural effusions may be present, and the involvement of lymph nodes, usually above the diaphragm, may be a prominent feature. Bone, skin, bone marrow, the central nervous system, and abdominal organs (but rarely bowel, as seen in BL) may also be involved, and occasionally other sites such as lymphoid tissue of Waldeyer ring and testes. 
As opposed to other NHLs in children, where intensive chemotherapy given over a period of less than 1 year yields excellent results, the best results in LL (including stage 1 and 2 disease) have been achieved with regimens designed to treat ALL. These include relatively intense induction and consolidation therapy, followed by 2 years of much less intense maintenance therapy.66 Expected cure rates for pediatric LL are 85% or greater.67 Outcome for patients with refractory or recurrent disease is similar to Burkitt lymphoma (BL), that is, less than 25% survival rate.
Recent attempts to intensify therapy have not improved the outcome for the majority of patients with LL. Therefore, new approaches in the diagnosis, pathogenesis, and treatment of children and adolescents with LL are focused on early identification of those likely to relapse. These individuals would be subjected to more aggressive and novel therapies, and therapy of patients with excellent prognosis could perhaps be reduced.
NON-HODGKIN LYMPHOMA (NHL) OF IMMUNODEFICIENCY
The incidence of lymphoproliferative disease or lymphoma is 100-fold higher in immunocompromised children than in the general population. It is estimated that as many as 100 to 150 cases of NHL in immunodeficient children and adolescents are observed in the United States per year, making this group of disorders as common as diffuse large B-cell lymphoma (DLBCL) or anaplastic large-cell lymphoma (ALCL). The immune deficiencies may be the result of a genetically inherited defect, HIV infection, or iatrogenic following transplantation [solid organ transplantation or allogeneic hematopoietic stem cell transplantation (HSCT)]. Epstein-Barr virus (EBV) is associated with most of these tumors, but some tumors are not associated with any infectious agent. 
Disease in immunocompromised patients rarely affects peripheral lymph nodes and extra-lymphatic sites are very common, including primary CNS lymphoma (PCNSL), which is rarely seen in children who are immunocompetent. Regardless of the immune defect, immunodeficient children with lymphoma have a worse prognosis than does the general population with non-Hodgkin lymphoma (NHL).69,73,74 If the disease is localized and amenable to complete surgical resection and/or radiation therapy, the outcome is quite favorable. However, most NHL in this population is disseminated and requires systemic cytotoxic therapy. These patients usually tolerate cytotoxic therapy poorly, with increased morbidity and mortality due to increased infectious complications and often increased end-organ toxicities. Highly active anti-retroviral therapy (HAART) has decreased the incidence of NHL in HIV-infected patients. Recent studies demonstrate that children with HIV and NHL should be treated with standard chemotherapy regimens for NHL, but careful attention to prophylaxis against and early detection of infection is warranted. Patients with primary immunodeficiency can achieve complete and durable remissions with standard chemotherapy regimens for NHL, though again toxicity is increased and correction of underlying immunologic defect through allogeneic hematopoietic stem cell transplantation (HSCT) is often required to prevent recurrences. In Postrans-plant lymphoproliferative disease (PTLD), first-line therapy is the reduction of immunosuppressive agents to doses as low as possible. Rituximab, an anti-B-cell antibody, has been used with some success, but data for its use in children are sparse. As many as 70% of children with EBV positive PTLD following organ transplant who had failed other therapies, can be cured with a low-dose chemotherapy regimen.73
 SUMMARY
SUMMARY
Through tailoring of therapy to specific subtypes and stages of disease, the cure rates for non-Hodgkin lymphoma (NHL) in children and adolescents is now 70% to 90%, even for patients with the most advanced disease. Although radiotherapy has now been largely eliminated from therapy, certainly short-term toxicities and potentially long-term complications remain problematic for these patients. The challenges for the future are (1) to identify which patients can maintain excellent outcomes with less therapy; (2) to identify patients who are at a high risk to relapse and intervene before relapse, as currently the outcome for these patients is dismal; and (3) to better understand the biology of all subtypes of NHL and develop more targeted, biologic therapies so cure rates can be improved with fewer complications.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


