Neurotransmitter Disorders
Erika Fullwood Augustine and Annapurna Poduri
The pediatric neurotransmitter disorders are a fairly recently described group of inherited neurometabolic disorders related to defects in neurotransmitter synthesis or breakdown. These disorders may be divided into defects of γ-aminobutyric acid (GABA), glycine or monoamine metabolism, and secondary neurotransmitter deficiency states. This chapter will primarily discuss disorders of monoamine metabolism; we will focus on the diseases best characterized to date, including GTP cyclohydrolase deficiency, tyrosine hydroxylase deficiency, aromatic L-amino acid decarboxylase deficiency, and monoamine A deficiency.
The monoamine neurotransmitters include biogenic amines or catecholamines (dopamine, norepinephrine, epinephrine) and serotonin. Synthesis of dopamine and serotonin begins with tyrosine and tryptophan. Tyrosine is hydroxylated by tyrosine hydroxylase and tryptophan by tryptophan hydroxylase to form levodopa and 5-hydroxytryptophan, respectively. Levodopa and 5-hydroxytryptophan are then catalyzed to dopamine and serotonin by aromatic L-amino acid decarboxylase (AADC). See Figure 577-1.
Dopamine is converted to norepinephrine via dopamine β-hydroxylase; norepinephrine is then metabolized to epinephrine. Dopamine and serotonin are also deaminated by monoamine oxidase A and B to homovanillic acid and 5-hydroindoleacetic acid, respectively, metabolites that can be measured in CSF. Dopamine is also broken down by catechol-o-methyltransferase to 3-O-methyldopa, also detectable in CSF. See Figure 577-1.
Tetrahydrobiopterin serves as a cofactor for both tyrosine hydroxylase and tryptophan hydroxylase. Similarly, pyridoxine (vitamin B6) is a cofactor for AADC. Thus, impairments in tetrahydrobiopterin or pyridoxine metabolism may result in deficiencies of monoamine neurotransmitters as well.
The disorders in the pathways described above typically affect the basal ganglia due to dopamine deficiency and thus present with hyper-or hypokinetic abnormalities of movement. However, there are widespread effects across the cerebrum causing a broad range of symptoms. Patients may present subtly with global developmental or motor delays, but the presence of abnormalities of tone such as spastic diplegia, dystonia, autonomic dysfunction, or evidence of disease progression should point toward this group of disorders. Deficits in neurotransmitter metabolism should be considered in the differential diagnosis of progressive encephalopathies, particularly those with onset in infancy. The diagnosis of these conditions is often made late due to the lack of awareness of this group of diseases.
GUANOSINE TRIPHOSPHATE CYCLOHYDROLASE DEFICIENCY
Guanosine triphosphate cyclohydrolase (GCH1) catalyzes the rate-limiting step in tetrahydrobiopterin synthesis, thus impairment of this enzyme results in deficiencies of serotonin and dopamine. GCH1 deficiency was first described by Segawa in 1971.1 Also known as Segawa disease, dopa-responsive dystonia, or hereditary progressive dystonia, the disorder is characterized by dystonia or tremor with diurnal fluctuation. Patients typically present in the first decade of life with gait abnormality due to dystonia of the lower extremities. Symptoms are worse later in the day and improved following sleep. Over the course of 10 to 15 years, dystonia gradually spreads to involve all 4 limbs. Postural tremor develops in the upper extremities and spreads to involve the head and neck. By adulthood, parkinsonian signs predominate. Symptoms become static, with less diurnal variation by the fourth decade. The predominant symptoms appear to be determined by a patient’s age, regardless of the age at onset or duration of disease.2 Short stature, sleep disturbances, and psychiatric issues, including increased risk of depression and obsessive-compulsive symptoms, have been reported in association with GCH1 deficiency.2,3
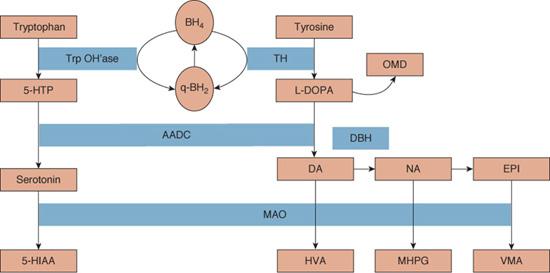
FIGURE 577-1. Synthesis and metabolism of monoamine neurotransmitters (from Pediatric Neurotransmitter Disorders, Pearl et al19). 5-HIAA, 5-hydroxyindoleacetic acid; 5-HT, 5-hydroxytryptophan; AADC, aromatic L-amino acid decarboxylase; BH2, dihydrobiopterin; BH4, tetrahydrobiopterin; GTP, GTP cyclohydrolase; HVA, homovanillic acid; L-dopa, levodopa; MAO, monoamine oxidase; MHPG, 3-methoxy-4-hydroxyphenylglycol; VMA, vanillylmandelic acid. BH4 is cofactor for tyrosine hydroxylase (TH) and tryptophan hydroxylase (Trp OH’ase).
Clinical examination reveals asymmetric dystonia, most prominent in the lower extremities initially. If the examination is early in the day, however, the dystonia may be minimal. Increased lower extremity tone, hyperreflexia, and extensor plantar responses may be present. Cognition remains normal throughout the clinical course.
GCH1 deficiency, as described above, is an autosomal dominant disorder with variable penetrance; over 100 mutations in the GCH1 gene (14q22.1-22.2) have been described. Considerable variability in phenotype has been observed with respect to symptom onset, clinical manifestations, and severity, even within the same family. Symptoms ranging from mild exertion-induced dystonia to severe dystonia and parkinsonism have been described.4 Patients may also present with tremor, spastic paraparesis, or infantile hypokinetic rigid syndromes.5 In sporadic mutations, there is a female-to-male ratio of 4:1; the literature supports a sex effect on phenotype as well, with more severe cases occurring in girls. Genetic testing is clinically available, although a known mutation is not identified in up to 40% to 50% percent of cases.6,7 Note that there is also a less common severe autosomal recessive form of GCH1 deficiency characterized by global developmental impairment, hypotonia, autonomic dysfunction, seizures, dystonia, and oculogyric crises.
CSF evaluation shows low homovanillic acid, low neopterin, and low biopterin concentrations. See Table 577-1 for comparison to other disorders.
The hallmark of the disorder is dramatic sustained responsiveness to low doses of levodopa-carbidopa, which are generally well tolerated. Note that other basal ganglia diseases, such as juvenile parkinsonism, may also respond to levodopa. However, onset in early childhood, diurnal fluctuation, appearance of tremor later in life, and preservation of cognition should help differentiate guanosine tri-phosphate cyclohydrolase deficiency from other disorders.8,9
TYROSINE HYDROXYLASE DEFICIENCY
Tyrosine hydroxylase catalyzes the conversion of tyrosine to levodopa, the rate-limiting step in the synthesis of catecholamines. Tyrosine hydroxylase deficiency typically presents as a progressive encephalopathy with extrapyramidal and autonomic symptoms. Onset of symptoms generally occurs in the first 2 years of life. Extrapyramidal manifestations include oculogyric crises, dystonia, parkinsonism, tremor, or choreoathetosis. Observed alterations in tone range from truncal hypotonia to rigidity and severe spasticity; even without frank parkinsonism, an overall paucity of movement is observed. The phenotypic spectrum is broad; patients may display infantile parkinsonism, spastic paraparesis, exercise-induced dystonia, or progressive gait disturbance as the main symptoms. In contrast to guanosine triphosphate cyclohydrolase (GCH1) deficiency, the age of onset is earlier, dystonia develops later in the clinical course, and diurnal fluctuation is a less prominent feature. Over the course of the disease, muscle tone increases, contractures develop, and psychomotor retardation and immobilization ensue.10 The encephalopathy most commonly manifests as irritability. Tyrosine hydroxylase deficiency may result in mild autonomic dysfunction due to norepinephrine and epinephrine deficiency, causing ptosis, miosis, postural hypotension, temperature instability, and excessive salivation.
In addition to dystonia, physical examination demonstrates truncal hypotonia, appendicular hypertonia, and hyperreflexia with extensor plantar responses.
CSF evaluation demonstrates low homovanillic acid (HVA) and low 3-methoxy-4-hydroxyphenylglycol levels, with normal 5-hydroindoleacetic acid, neopterin, and biopterin. Plasma catecholamine levels are normal, although urine HVA is decreased. See Table 577-1 for comparison to other disorders. Gene sequencing is also available for clinical testing.
Table 577-1. Laboratory Studies in Selected Monoamine Disorders
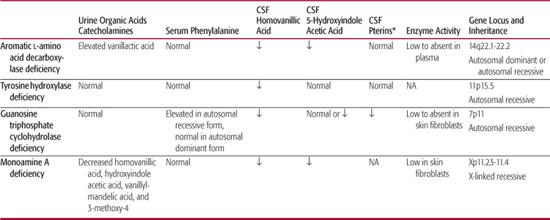
*Pterins, neopterin and biopterin.
NA, data not available.
Often referred to as an autosomal recessive dopa-responsive dystonia, some patients do respond to treatment with dopamine. However, many have incomplete or absent response to levodopa. Those who present with infantile parkinsonism are the least likely to benefit and tend to have the highest incidence of side effects related to levodopa administration.11 Levodopa appears to be most helpful for later-onset forms, but may take weeks to months to produce an effect, in contrast to GCH1 deficiency, for which benefit is typically observed within days.10 In tyrosine hydroxylase deficiency, prognosis overall remains poor.
AROMATIC L-AMINO ACID DECARBOXYLASE DEFICIENCY
Aromatic L-amino acid decarboxylase (AADC) catalyzes the conversion of levodopa to dopamine and 5-hydroxytryptophan to serotonin; defects result in dopamine, norepinephrine, epinephrine, and serotonin deficiencies. First described in 1990, this rare autosomal recessive disorder is characterized by global developmental delay, extrapyramidal symptoms, hypotonia, and severe autonomic dysfunction.12,13 Most patients present in the first 6 months of life, with approximately 50% demonstrating symptoms in the neonatal period.14 Neonatal presentation is characterized by failure to thrive, with poor suck, swallowing dysfunction, hypothermia, lethargy, ptosis, and hypotension. Due to presentation with bradykinesia and athetosis, dystonic cerebral palsy is a common initial diagnosis.13,14 Dystonia, tremor, hypokinesia, and oculogyric crises compose the movement disorder. Truncal hypotonia and hypokinesia can be profound, although deep tendon reflexes are preserved. Limb hypertonia and emotional lability are common, and variable degrees of cognitive impairment are observed.
Of the disorders described in this chapter, AADC deficiency has the most clinically significant autonomic dysfunction, including paroxysmal sweating, hypotension, bradycardia, and hypothermia. The autonomic dysfunction is of particular importance in patients who require anesthesia for procedures or surgeries. Parasympathetic outflow is intact, uncountered by appropriate sympathetic response to stress. Compensatory responses to hypovolemia or blood loss are poor, and patients are at risk for severe bradycardia, refractory hypotension, and apneic episodes with administration of anesthesia or even relatively mild sedation.15
As seen in guanosine triphosphate cyclohydrolase deficiency, there appears to be a divergence of male and female phenotypes. Males tend to demonstrate a better response to treatment, whereas females suffer from frequent adverse treatment effects and experience only minimal or transient treatment response.16
CSF studies show decreased homovanillic acid, 5-hydroindoleacetic acid, and 3-methoxy-4-hydroxyphenylglycol, and elevated levodopa, 5-hydroxytryptophan, and 3-O-methyldopa levels. CSF neopterin and biopterin are normal. Urine organic acid testing shows elevated vanillactic acid, normally absent from urine; hyperdopaminuria from intact kidney AADC may also be present.17 A plasma AADC enzyme assay is also available. See Table 577-1 for comparison to other disorders. Brain MRI is generally unremarkable, but may show progressive cerebral atrophy.14 Gene sequencing is available for clinical testing.
Treatment is very limited, mostly impacting the movement disorder, although patients may demonstrate small benefit from dopamine agonists, monoamine oxidase inhibitors, and vitamin B6 (pyridoxal-5-phosphate is a cofactor for AADC).
MONAMINE A DEFICIENCY
Monoamine oxidase A and B (MAO-A and MAO-B) are mitochondrial enzymes that catalyze the deamination of biogenic amines. MAO-A is found mainly in fibroblasts in which its main effects are on the breakdown of serotonin and norepinephrine. A rare X-linked disorder, MAO-A deficiency presents in males with mild mental retardation, aggression, and violent behaviors. In a case series of 14 males with an MAO-A gene mutation, episodes of violent behaviors clustered into 1- to 3-day periods, with decreased sleep and night terrors.18 Measurement of plasma 3-methoxy-4-hydroxyphenylglycol (MHPG) can be used as an index of MAO-A activity; enzyme activity can also be measured in skin fibroblasts.11,19 Urine studies show decreased homovanillic acid, 5-hydroindoleacetic acid, MHPG, and vanillylmandelic acid.18 See Table 577-1 for comparison to other disorders.
DISORDERS OF γ–AMINO BUTYRIC ACID METABOLISM
Glutamic acid decarboxylase (GAD) converts glutamate to γ–amino butyric acid (GABA). Due to the role of pyridoxal-5-phosphate as a cofactor for GAD, pyridoxine-dependent seizures—presenting typically as refractory epilepsy—were previously thought to be related to GAD deficiency, but thus far the only gene associated with pyridoxine-dependent seizures is ALDH7A1.20 Clinical characteristics of true GAD deficiency are not yet well characterized, but it has been observed that the GAD67 gene and GABA may play a role in facial and palate development.21
Succinic semialdehyde dehydrogenase (SSADH) deficiency, also referred to as 4-hydroxybutyric aciduria, is an autosomal recessive disorder of GABA catabolism. SSADH catalyzes the conversion of succinic semialdehyde to succinic acid. In the absence or deficiency of SSADH, succinic semialdehyde is converted to γ-hydroxybutyrate, as depicted in Figure 577-2. The most frequent clinical features include mental retardation, language delay, hypotonia, and motor delay. Seizures occur in approximately one half of patients, and ataxia is nonprogressive, with isolated reports of improvement with age. A number of ocular abnormalities have been reported, including strabismus and nystagmus.22 Behavioral difficulties are reported in about 30% of patients, including anxiety, aggression, hyper-activity, hallucinations, sleep disturbance, or psychosis.23,24 In contrast to previously discussed neurotransmitter disorders, clinical manifestations do not include encephalopathy. MRI most commonly shows abnormal T2 signal of the subcortical white matter and globus pallidus. This disorder is diagnosed via urine organic acid analysis, which reveals elevated γ-hydroxybutyric acid (4-hydroxybutyric acid); metabolic acidosis is absent. SSADH enzyme activity can also be measured in leukocytes. Treatments with vigabatrin and benzodiazepines have had mixed results. SSADH should be considered during the evaluation of patients presenting with mental retardation and/or ataxia with a history of epilepsy, especially in the setting of consanguinity. (See Chapter 160.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


