Neuroblastoma
Michael D. Hogarty and Garrett M. Brodeur
Neuroblastoma, a tumor of the sympathetic nervous system, is the most common solid tumor in childhood. Interestingly, some infants with metastatic disease experience complete tumor regression without therapy, and other patients may have maturation of their tumor into a benign ganglioneuroma. Nevertheless, the majority of patients have metastatic disease at diagnosis that progresses despite intensive multimodality therapy.1-3 Current risk classification schemes use biological and clinical features at diagnosis to predict tumor behavior and to stratify patients to an appropriate treatment. Children with tumors that have lower risk features are spared unnecessary therapies yet still achieve excellent outcomes. However, the survival of patients with high-risk neuroblastoma is still unacceptably low. Advances in understanding the molecular pathogenesis of this tumor, including how alterations in specific biological pathways impact tumor behavior, may lead to novel therapeutics to reduce toxicity in patients with favorable disease and improve outcomes in those with unfavorable disease.4,5
 EPIDEMIOLOGY, GENETICS, AND MOLECULAR PATHOGENESIS
EPIDEMIOLOGY, GENETICS, AND MOLECULAR PATHOGENESIS
Neuroblastoma is the most common malignancy diagnosed in infants and accounts for 8% to 10% of childhood cancers overall. Unfortunately, it also accounts for 15% of childhood cancer-related deaths. The prevalence is about 1 per 7000 live births, and there are about 650 new cases per year in the United States, with an incidence of 10.5 per million per year in white and 8.8 per million per year in black children less than 15 years of age.1-3 This incidence appears fairly uniform throughout the world. The tumor is slightly more common in males than in females, with a male-to-female ratio of 1.2:1 in most large studies. The median age at diagnosis is 22 months, and less than 5% of patients are diagnosed after 10 years of age. The rare adolescent or young adult with neuroblastoma poses a unique treatment challenge, as their tumors appear biologically distinct with an indolent and often progressive course.
The etiology of neuroblastoma is unknown in most cases, but it appears unlikely that environmental exposures play a major role, because no prenatal or postnatal drug, chemical, or radiation exposure has been strongly or consistently associated with an increased risk. Neuroblastoma has been reported in patients with neurofibromatosis type 1, as well as central congenital hypoventilation syndrome (CCHS) and Hirschsprung disease, suggesting that disordered neural crest development may predispose to neuroblastoma. Mutations in the PHOX2B gene, a key regulator of autonomic neural development, have been identified in neuroblastomas associated with these latter disorders, and in occasional sporadic tumors.6,7 Diverse congenital anomalies have also been reported in association with neuroblastoma, but without a causal genetic etiology. Finally, there may be a decreased prevalence of neuroblastoma in patients with Down or Klinefelter syndromes, and an increase in Turner syndrome, but the reasons for this are unclear.
As with many embryonal tumors, a small subset of neuroblastoma patients has a familial predisposition that follows an autosomal dominant pattern of inheritance. Analysis of such pedigrees predicted that neuroblastoma fits the 2-mutation hypothesis proposed by Knudson for the origin of childhood cancers. The median age at diagnosis is younger (9 months in contrast with 22 months), and multiple primary tumors are more prevalent in familial cases. Heritable mutations within the ALK gene, an oncogenic tyrosine kinase deregulated in multiple human cancers, is causative in the majority of familial cases, with gene duplication or amplification believed to act as the “second hit.”8 Acquired mutations in ALK also play a role in a subset of sporadic neuroblastomas, providing a potential therapeutic target.9
Trk Receptors, Differentiation, and Regression
Neuroblastoma arises from sympathetic neuro-blasts and often demonstrates neuronal differentiation. Some tumors undergo spontaneous or therapy-induced differentiation to ganglioneuroma, so the malignant behavior of these cells may be maintained in part by a failed differentiation program. Additionally, some neuroblastomas regress spontaneously, particularly in infants. The factors responsible for regulating differentiation or regression in these tumors are not well understood but may involve neurotrophin signaling, as these pathways are paramount in modeling the sympathetic nervous system. Three neurotrophin receptors (NTRK1, NTRK2, and NTRK3 encoding TrkA, TrkB, and TrkC) were identified. Their primary ligands are nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin-3 (NT-3), respectively. 
Overall, the pattern of expression of the neurotrophin receptors may play an important role in the behavior of neuroblastomas, including the likelihood that a tumor may undergo spontaneous regression or differentiation. 
Genomic Classification of Neuroblastomas
Several distinct genomic changes have been identified as characteristic of neuroblastoma subsets. Amplification of the MYCN protooncogene is seen in 20% to 25% of tumors and strongly associates with unfavorable disease outcome.12 Testing for this genomic alteration is recommended for all neuroblastomas, as these results are used for treatment stratification worldwide.4,5 In addition, common regions of deletion or allelic loss involving 1p or 11q (among other loci) and unbalanced gain of distal 17q have been associated with advanced disease stage and poor outcome in numerous studies.13-16CHD5 has been identified as a tumor suppressor gene deleted from 1p36.31, but the cancer-associated genes from the other regions have yet to be identified.17 Diploid or tetraploid DNA content has also been recognized as a feature of unfavorable neuroblastomas.18 In contrast, tumors with hyperdiploid or near triploid DNA content with whole chromosome gains have a more favorable outcome.
More recently, technologies capable of identifying genome-wide copy-number alterations in tumor cells have been applied to neuroblastoma. These data suggest that there appear to be three distinct genomic subsets of neuroblastoma that are highly correlated with clinical outcome.4,5 The first is characterized by mitotic dysfunction leading to a hyperdiploid modal karyotype, with whole chromosome gains but few if any structural chromosome rearrangements (Fig. 457-1). These patients are generally infants or toddlers with localized disease and a very good prognosis. The majority of tumors, however, are characterized by a near-diploid or tetraploid karyotype with segmental chromosomal aberrations, and unbalanced 17q gain is found in the majority. Within this less favorable genomic type, two subsets can be distinguished. One demonstrates segmental chromosomal changes, such as 11q or 3p deletion. The other is defined by high-level amplification of the MYCN proto-oncogene, frequently with 1p deletion. These latter tumors are highly aggressive and often lethal. Patients with these unfavorable subtypes are generally older than 1 year with more advanced stages of disease, which is often progressive. Current evidence suggests that these are genomically and biologically distinct, and that there is no progression from one genomic subtype to another. The underlying causes of the genomic instability that lead to these patterns of genomic change remain unknown.
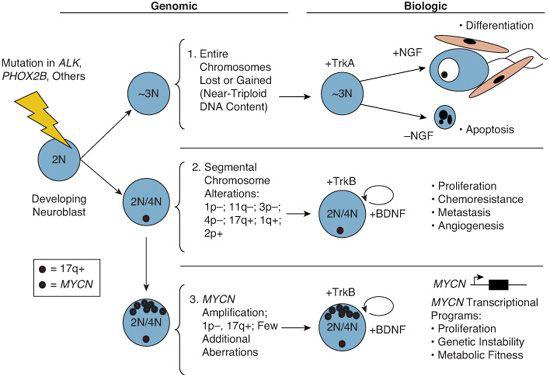
FIGURE 457-1. Genomic and biologic classification of neuroblastoma. Neuroblastomas are derived from sympathetic neuroblasts in the developing nervous system that undergo select genetic alterations (acquired or germline). Subset 1 is characterized by hyperdiploidy or near-triploidy with gains and losses of apparently intact chromosomes. These tumors rarely demonstrate high-risk tumor features and have favorable outcomes. Most express TrkA and may activate differentiation or apoptosis programs depending on the presence of its ligand (NGF) in their microenvironment. In contrast, the majority of neuroblastomas demonstrate segmental structural chromosome alterations (subsets 2 and 3) with loss or gain of genetic material that presumably results in suppressor gene inactivation or oncogene activation, respectively. Unbalanced gain of 17q is common to many of these. Subset 2 may also have one or several other nonrandom chromosome aberrations, such as 1p, 3p, 4p, or 11q deletion, or gain of 1q or 2q. Subset 3 is usually near-diploid or near-tetraploid with high-level amplification of a region of chromosome 2p harboring the MYCN proto-oncogene. Most tumors with MYCN amplification also have 1p deletions with loss of one copy of CHD5. MYCN-amplification and CHD5 deletion are markers of highly aggressive disease, correlating with numerous unfavorable clinical and biological features as well as a poor outcome.
 CLINICAL FEATURES
CLINICAL FEATURES
Primary Tumors
About half of all neuroblastomas originate in the adrenal medulla; 30% occur in nonadrenal abdominal sites in the paravertebral ganglia, pelvic ganglia, or the organ of Zuckerkandl; and 20% occur in the paravertebral ganglia of the chest or neck.1 Most primary tumors cause symptoms such as abdominal mass or pain. Paraspinal tumors frequently invade the spinal canal through neural foramina and may cause spinal cord compression. In the thoracic or upper lumbar region, this may lead to paraplegia, whereas lower lumbar invasion leads to a cauda equina syndrome with loss of bowel or bladder function. These are true oncologic emergencies, and rapid institution of therapy may be necessary to preserve neurologic function. Midline tumors can displace or compress structures such as the trachea or esophagus and lead to obstructive symptoms. Involvement of the superior cervical ganglion can produce Horner syndrome as its initial manifestation.
Metastases
More than half of all neuroblastomas are meta-static at the time of diagnosis. Frequent sites of metastasis are regional or distant lymph nodes, cortical bone, bone marrow, and liver. In infants, there is a characteristic pattern of metastases which shows massive infiltration into the liver, skin nodules, and variable bone marrow involvement. This pattern of disease is referred to as stage 4S disease using the International Neuroblastoma Staging System, and it has a very favorable outcome.19 However, in older patients (≥ 18 months old), metastasis frequently involves significant bone marrow and cortical bone dissemination, commonly including the skull and orbits. Rarely, disease may spread to lung and brain parenchyma, usually as a manifestation of relapsing or end-stage disease. The outlook for patients such as these is poor, even with intensive, multimodality therapy.
Paraneoplastic Syndromes
Several paraneoplastic syndromes have been rarely associated with neuroblastoma, representing only 1% to 3% of patients.1
1. Vasoactive intestinal peptide (VIP) syndrome. These patients have intractable secretory diarrhea and abdominal distention, sometimes associated with hypokalemia and dehydration, which is a manifestation of tumor secretion of VIP. The biological functions of VIP are relaxation of smooth muscle, stimulation of intestinal water and electrolyte secretion, and stimulation of release of other polypeptide hormones. The VIP syndrome usually is associated with ganglioneuroblastoma or ganglioneuroma, and these symptoms usually resolve after eradication of the tumor.
2. Opsomyoclonus-myoclonus-ataxia (OMA) syndrome. This syndrome consists of myoclonic jerking and random, uncontrolled eye movement, sometimes associated with cerebellar ataxia. These patients usually have biologically favorable tumors and an excellent tumor outcome with surgery alone. Unfortunately, many have persistent severe neurologic abnormalities. OMA syndrome may be caused by antineuronal autoanti-bodies. The symptoms vary in severity, worsening with intercurrent illnesses, and may respond to immune modulation. Chemotherapy treatment may also improve the neurologic outcomes by more rapidly eliminating antigenic tumor cells, as well as providing immune suppression.
3. Excessive catecholamine secretion syndrome. Rarely, neuroblastoma patients may present with tachycardia, hypertension, palpitations, profuse sweating, and flushing. This syndrome is more common in patients with pheochromocytomas; neuroblastoma patients with hypertension most often have a renovascular etiology, rather than catecholamine secretion.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Because of its myriad clinical presentations, neuroblastoma may be confused with other neoplastic and nonneoplastic conditions. This may be a problem particularly in the 5% to 10% of patients with tumors that do not produce excess catecholamines, as well as the 1% who do not have an obvious primary tumor. The VIP syndrome can be confused with inflammatory bowel disease, and those with opsoclonus-myoclonus and ataxia syndrome can resemble a primary neurologic disorder. Histologically, neuroblastoma tissue from primary or meta-static sites may be quite undifferentiated and may be confused with other embryonal pediatric cancers such as rhabdomyosarcoma, Ewing sarcoma, lymphoma, or even leukemia (especially megakaryoblastic leukemia). Molecular, biochemical, and immunological characterization can usually distinguish among these various possibilities.
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Diagnosis
To confirm a diagnosis of neuroblastoma, characteristic histological or ultrastructural features are sought using light microscopy, immunohistochemistry, and rarely electron microscopy. Stratification to an appropriate therapy often requires that tumor genomic features and histopathologic grade be known, so sufficient tumor material should be obtained in these cases. Some patients with neuroblastoma and metastatic disease are diagnosed based on the presence of tumor cells within the bone marrow accompanied by increased urinary catecholamine metabolites.
The majority of neuroblastomas (greater than 90%) produce sufficient catecholamines to result in increased urinary metabolites.1 This provides an ancillary diagnostic test, as well as a means to follow disease activity, including surveillance for relapse. The two enzymes primarily responsible for the catabolism of catecholamines are catechol-O-methyl transferase and monamine oxidase. DOPA and dopamine are converted primarily to homovanillic acid (HVA), whereas norepinephrine and epinephrine are converted primarily to vanillylmandelic acid (VMA). Both urinary VMA and HVA should be normalized for age and for urinary creatinine.
Evaluation
A standard set of recommended tests to define the clinical stage or extent of disease has been established. Computerized tomography (CT) scan is the current preferred imaging modality for primary tumors, although concerns regarding radiation exposure in young children may lead to more frequent use of magnetic resonance imaging (MRI) scan. MRI is superior in evaluating paraspinal mass lesions for intraforaminal extension and spinal cord compression. 123 I-Meta-iodobenzylguanidine scintigraphy (MIBG scan) is used for evaluation of the primary tumor and metastatic disease sites. MIBG is selectively concentrated by catecholaminergic cells, including greater than 90% of neuroblastomas, providing highly sensitive and specific detection of metastases. If the primary tumor does not take up MIBG (as in 5% to 10% of neuroblastomas), then a 99 mTc-diphosphonate scintigraphy (bone scan) is recommended to assess for metastatic cortical bone sites. The use of positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose (FDG) is being evaluated alone and in combination with CT (PET-CT) for its utility in staging and monitoring disease status.
Bone marrow disease is assessed via bilateral bone marrow aspirates and biopsies with standard histological analysis. Molecular or immunocytochemical assays that detect neuroblastoma cells in marrow or blood samples at diagnosis and following treatment, have also been developed, to assess minimal residual disease. Although there is no doubt that these techniques increase the sensitivity for detecting neuroblastoma cells by 1 or 2 orders of magnitude, it is not yet clear if this level of sensitivity provides information that is useful clinically. They are currently not used for assessing bone marrow involvement, which instead relies on visual detection of neuroblast clumps by light microscopy.
Tumor Histology
Neuroblastomas arise from primitive pluripotential sympathetic nerve cells (sympathogonia) that are derived from the neural crest. These cells can differentiate into the normal tissues of the sympathetic nervous system, such as the spinal sympathetic ganglia, the supporting Schwannian cells, and adrenal chromaffin cells. Neuroblastic tumor histopathologies include neuroblastoma, ganglioneuroblastoma, and ganglioneuroma, reflecting increased neural differentiation. The typical neuroblastoma is composed of small cells containing dense, hyperchromatic nuclei and scant cytoplasm. The presence of neuritic processes, or neuropil, is a pathognomonic feature of all but the most primitive neuroblastoma. The fully differentiated, benign counterpart of neuroblastoma is ganglioneuroma, which is composed of mature ganglion cells, surrounded by a matrix of Schwannian cells and neuropil. Ganglioneuroblastomas have intermediate maturation and may be focal or diffuse or have nodules of malignant neuroblastoma within an otherwise mature ganglioneuroma (nodular ganglioneuroblastoma). Shimada and colleagues devised a classification schema relating histopathologic tumor features to clinical outcome. Tumors are classified as favorable or unfavorable depending upon the degree of neuroblast differentiation, Schwannian stroma content, mitosis-karyorrhexis index (MKI), and age at diagnosis. A modification of this system, the International Neuroblastoma Pathology Classification System (INPC), is currently used,20 but future systems will define histo-logical features independent of patient age.
Staging
To provide a common staging system for comparison of clinical trial results worldwide, the International Neuroblastoma Staging System (INSS) was developed.18 In addition to using image-defined tumor features (such as degree of locoregional or distant spread), this system depends on surgical factors, such as completeness of initial resection, that may differ from one institution to another and lead to differences in stage assignment. A presurgical International Neuroblastoma Risk Group Staging System (INRGSS) has been proposed in which only the radiographic characteristics of the tumor, as well as bone marrow morphology, are used to more uniformly define extent of disease at diagnosis.21 This staging system will distinguish locoregional tumors that do not involve local structures from locally invasive tumors. Metastatic tumors will remain similar to current INSS stage 4 and 4S patterns of disease, respectively.
 PROGNOSTIC CONSIDERATIONS
PROGNOSTIC CONSIDERATIONS
Clinical Variables
Variables shown to have prognostic significance in neuroblastoma are collectively used to stratify patients to an appropriate treatment. The most important clinical variables are stage of disease (see above) and the age of the patient at diagnosis. Localized tumors (stage 1 and 2) are more common in younger children and are associated with favorable outcomes. Young children with stage 4S disease, a unique pattern of dissemination, also have favorable outcomes despite the presence of metastases. In contrast, older children more commonly have tumors that are regionally infiltrative (stage 3) or disseminated (stage 4) and have a poor overall survival. Although younger age and lower stage of tumor are associated with good outcomes in general, a subset of these tumors manifests a very aggressive clinical course. These tumors frequently have unfavorable biological features, underscoring the importance of using both clinical and biological information (see below) for risk assessment. Younger patient age is independently predictive of more favorable outcome in neuroblastoma. Age is likely a surrogate for developmental changes occurring in the neuroblasts that become cancerous or in the cancer microenvironment, and age may be considered as a continuous variable. For obvious practical reasons, however, age has traditionally been analyzed as a binary variable with < 365 days defined as favorable. An analysis of more than 3000 patients with neuroblastoma suggests that this value may be too low, and 547 days (∼18 months) may be more appropriate.22
Biological Variables
Tumor histopathology (discussed above), serum markers, and select genomic features also have independent prognostic value. Serum levels of ferritin, neuron-specific enolase, and LDH may correlate with disease burden and outcome, but they have largely been supplanted by more tumor-specific biological variables. The DNA index (reflecting tumor cell DNA content, or ploidy) is a strong prognostic marker, particularly for younger patients with disseminated disease. Hyperdiploid (often near-triploid) DNA content correlates with favorable disease behavior, whereas near-diploid (and near-tetraploid) tumors tend to be more aggressive, but this correlation loses its significance after 18 to 24 months of age. The specific genomic aberration most consistently associated with poor outcome in neuroblastoma is amplification of the MYCN proto-oncogene, which occurs in ∼20% of tumors and is strongly correlated with a poor outcome, even in patients with favorable age and stage. Deletion of 1p is found in ∼35% of neuroblastomas and correlates with MYCN amplification and advanced disease stage. 1p loss predicts for an increased risk of relapse in patients with localized tumors. Allelic loss of 11q is also present in ∼30% of tumors. Unlike 1p loss, this aberration is rarely seen in tumors with MYCN amplification, yet remains highly associated with other high-risk features suggesting it may be a biomarker of aggressive disease independent of MYCN pathways. A gain of 1 to 3 additional 17q copies, often through unbalanced translocation, may correlate with an aggressive phenotype. The presumptive cancer genes deregulated by most of these genomic changes remain unknown, and their prognostic significance relative to other genomic and biological markers are being further evaluated.
 TREATMENT AND OUTCOME
TREATMENT AND OUTCOME
The current Children’s Oncology Group (COG) risk stratification system incorporates patient age and INSS stage at diagnosis, as well as tumor histopathology, DNA index, and MYCN gene status to assign patients to 1 of 3 risk groups (low-, intermediate-, or high-risk) and to stratify treatment intensity accordingly. The goal of risk-adapted therapy is to maintain outstanding outcomes with minimal toxicity for children with favorable disease features while ensuring that children with unfavorable high-risk tumors are identified and receive intensive multimodality treatments, offering them the best chance of cure.
Neuroblastoma treatment may include surgery, chemotherapy, radiotherapy, and immunotherapy in high-risk cases, or observation alone in carefully selected low-risk cases. A comprehensive summary is beyond the scope of this review. However, a simplified approach requires that 2 critical issues be addressed. First, a determination must be made as to whether residual tumor remains following the initial surgical procedure. If so, integration of biologic and clinical data is crucial to predict the behavior of this residual tumor. Neuroblastoma has a remarkable propensity to regress or differentiate. Therefore, in some cases, the presence of macroscopic residual tumor, even at metastatic sites, is not an indication for adjuvant therapy. Conversely, some localized tumors can be near-totally resected, yet the presence of unfavorable biological features supports the need for adjuvant therapy.
Surgery
Surgery plays a pivotal role in the management of neuroblastoma. The goals of a primary surgical procedure are to provide tissue to establish the diagnosis and perform biomarker testing, and to excise the tumor if feasible without undue surgical risk. The majority of localized neuroblastomas have favorable biological features and can be successfully treated with surgery alone. In patients with unresectable tumors who require chemotherapy, subsequent delayed or second-look surgery may remove residual disease and facilitate histological assessment of treatment response.
Chemotherapy
Chemotherapy is the mainstay of therapy for neuroblastomas with unfavorable biological features. Phase I and II clinical trials conducted in patients with neuroblastoma have identified a number of effective drugs. Cyclophosphamide, ifosphamide, cisplatin, carboplatin, doxorubicin, and etoposide (VP-16) yield complete and partial response rates of 25% to 50% and have become the cornerstone of multiagent regimens. Newer effective agents include topotecan, irinotecan, and temozolamide. Drug combinations have been developed that take advantage of drug synergism, mechanisms of cytotoxicity, and differences in side effects. Treatment of children with advanced stage neuroblastoma using dose-intensive chemotherapy combinations, including myeloablative consolidation therapy with autologous stem cell rescue, has resulted in improved outcomes, but disease relapse and toxicities remain a significant problem. Newer agents under evaluation include drugs targeting key biological pathways supporting tumor progression and include angiogenesis inhibitors, retinoids, proapoptotic agents, immunotherapy (such as antibodies targeting the neural GD2 cell-surface marker), and kinase inhibitors.
Radiotherapy
Neuroblastoma is a radiosensitive tumor, and radiotherapy is effective in achieving local disease control or symptom palliation. However, long-term control of neuroblastoma is seldom achieved with radiation therapy alone because of the propensity of this tumor to metastasize. Historically, radiation has been used in the multimodality management of residual neuroblastoma and bulky unresectable tumors. The availability of effective chemotherapy regimens, concern over late toxicities of radiation exposure to developing tissues, and more accurate risk stratification have led to a marked reduction in radiotherapy use. Still, it is an important adjuvant therapy in high-risk disease to control against tumor relapse at sites of bulk disease. There is growing experience with targeted radiotherapeutic approaches such as131 Imetaiodobenzylguanidine (MIBG) to deliver a radiotherapy dose selectively to tumor cells. This agent has demonstrated potent activity against relapsed or refractory neuroblastoma.
Myeloablative Therapy with Hematopoietic Stem Cell Rescue
Attempts have been made to improve on the modest gains of intensive, combined-modality therapy by increasing the intensity of therapy. More intensive therapy can be administered if accompanied by hematopoietic stem cell transplant (SCT). Allogeneic SCT appears to offer little if any advantage and greater toxicity compared to autologous BMT. Currently, most centers use peripheral blood stem cells as the source of progenitor cells for marrow rescue following myeloablative conditioning. Although stem cell product contamination with neuroblasts is possible, studies have not demonstrated any benefit to efforts to remove these using monoclonal antibodies against tumor cells to purge the final stem cell product if gross contamination is not apparent. Thus, recurrence largely stems from neuroblasts not eradicated by high-dose cytotoxic therapy rather than through the reinfusion of viable cancer cells in the stem cell product.
The use of dose-intensified chemotherapy combinations, with or without autologous stem cell rescue, has improved immediate disease control in neuroblastoma. Unfortunately, this has not translated into durable remissions in the majority of children with high-risk tumors. Biological therapy to treat persistent minimal residual disease has been added following SCT. The use of 13-cis-retinoic acid, which induces neuroblast differentiation, in the posttransplant setting was tested in a randomized Phase III trial and showed improved event-free survival with acceptable toxicity.23 Thus, retinoid-based biotherapy in the post-transplant setting is now widely used. Other compounds, including novel retinoids such as Fenretinide, or immunotherapies, such as anti-GD2 antibody, have activity against minimal residual disease in high-risk neuroblastoma patients and warrant further study.
 FUTURE CONSIDERATIONS
FUTURE CONSIDERATIONS
Advances have been made in understanding the biology underlying neuroblastoma initiation and progression. However, there remain a variety of areas through which improvements in outcomes may be realized. These include (1) the ongoing identification of the principal genomic mutations driving tumor propagation (both germline and somatically acquired) that will inform our biological understanding and provide novel therapeutic targets, such as ALK; (2) continued refinements to biological classification (such as implementation of array-based technologies for biomarker determination) and risk-stratification schemas; and (3) the development of rational biologically based therapy targeted to the genes, proteins, and pathways responsible for initiation or maintenance of the malignant state.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


