Neuroblastoma
Neuroblastoma is the most common solid extracranial malignancy of childhood and the most common malignant tumor in infants.1 The overall incidence of neuroblastoma is 1 per 100,000 children in the USA, thereby comprising 7–10% of all malignancies diagnosed in patients younger than 15 years of age. Yet neuroblastoma is responsible for approximately 15% of all pediatric cancer deaths.2 Neuroblastoma is a heterogeneous disease; tumors can spontaneously regress or mature, or display a very aggressive, malignant phenotype.3 Because of these unique characteristics, neuroblastoma has been of great interest to both clinicians and basic science researchers. Progress in molecular and cellular biology in the past 30 years has contributed greatly to a better understanding of this disease. Unfortunately, this progress has not significantly altered the clinical outcome for patients with advanced-stage neuroblastoma. Although the prognosis for these patients has improved in the past three decades, the long-term outcome remains very poor.
The etiology of neuroblastoma is currently unknown, and no environmental factors have been convincingly linked to its development. The disease generally occurs sporadically, but familial neuroblastoma occurs in about 2% of the cases. The substantial biologic and clinical heterogeneity is also observed in familial cases.4 The germline mutation associated with hereditary neuroblastoma has been identified: activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) oncogene on the short arm of chromosome 2 (2p23).5 These mutations can also be somatically acquired, although the prevalence of ALK activation in sporadic neuroblastoma remains to be determined.
Pathology
Neuroblastoma is an embryonal tumor of the sympathetic nervous system. These tumors arise during fetal or early postnatal life from sympathetic cells (sympathogonia) derived from the neural crest. Therefore, tumors can originate anywhere along the path which neural crest cells migrate, including the adrenal medulla, paraspinal sympathetic ganglia, and sympathetic paraganglia such as the organ of Zuckerkandl. The German pathologist Rudolph Virchow is generally credited with being the first to describe the histologic appearance of what is now known as neuroblastoma in his 1864 article entitled, ‘Hyperplasia of the pineal and suprarenal glands’.6 The first to use the term ‘neuroblastoma’ was James Homer Wright in 1910 who described the classic appearance of rosettes of tumor cells around central neural fibrils.7 He also noted the association between the common sites of tumor development and the pattern of migration of primitive neural cells.
As one of the ‘small, round blue cell’ tumors of infancy and childhood, neuroblastoma, particularly when undifferentiated, must be distinguished from other neoplasms in this group (Ewing sarcoma family of tumors [ESFT], non-Hodgkin lymphoma, and rhabdomyosarcoma). Neuroblastoma can be distinguished histologically by the presence of neuritic processes (neuropil) and Homer Wright rosettes (neuroblasts surrounding eosinophilic neuropil). Scattered ganglion cells or immature chromaffin cells can also be seen. The appearance of the tumor cells may vary from undifferentiated cells to fully mature ganglion cells. In addition, neuroblastomas have variable degrees of Schwannian cell stroma, reactive non-neoplastic tissue recruited by the tumor cells. This stroma is intermixed, to a greater or lesser degree, as wavy bundles and sheets of spindle cells and produces anti-proliferative and differentiation-inducing factors that are crucial to neuronal differentiation.8,9 In addition, the Schwannian stroma appears to produce a variety of antiangiogenic factors, including pigment epithelium derived factor (PEDF)10 and secreted protein acidic and rich in cysteine (SPARC).11 Histopathologic variables among neuroblastic tumors include the degree of differentiation, maturation, lymphoid infiltration, calcification, anaplasia, necrosis, mitotic activity, neurofibrillary material (neuropil), and the presence of multinucleate cells. Finally, immunohistochemical analysis usually generates positive staining when antibodies to neuroblastoma-specific antigens such as synaptophysin, neuron-specific enolase, and chromogranin are used, and is negative when antibodies to actin, desmin, cytokeritin, leukocyte common antigen, vimentin, and CD99 are used.
Neuroblastoma is characterized by several unique clinical behaviors, including the secretion of catecholamine products and the potential to regress or mature, either spontaneously or in response to treatment. Small nodules of primitive neuroblasts are routinely found in the developing adrenal gland, even during the early postnatal period. Beckwith and Perrin described microscopic nodules that they termed ‘neuroblastoma in situ’ in the adrenals of infants undergoing autopsy following death from non-malignancy related causes.12 The incidence of this finding was more than 200-fold greater than the clinical incidence of neuroblastoma, which suggests that perhaps many neuroblastomas spontaneously regress or mature into lesions that never become clinically apparent. The process of involution is well described during embryonic life, especially in the developing central and peripheral nervous systems. Although initially thought to be mediated by the immune system, the process of involution may be the result of the withdrawal of neurotrophic maintenance factors such as nerve growth factor (NGF). Clinically apparent neuroblastoma can also regress or spontaneously mature, but the mechanism remains unknown.
Histopathologic Classification
In 1984, Shimada and colleagues first developed an age-linked classification system of neuroblastic tumors based on tumor morphology in which neuroblastomas were divided into two prognostic subgroups, favorable histology and unfavorable histology.13 In 1999, the International Neuroblastoma Pathology Classification (INPC) was devised, and then modified in 2003, and is an adaptation of the original Shimada system.14,15 The INPC is based mainly on morphologic changes associated with the maturational sequence of neuroblastic tumors. It remains an age-linked classification that depends on the differentiation grade of the neuroblasts, the cellular turnover index (mitosis-karyorrhexis index [MKI]), and the presence or absence of Schwannian stroma. The INPC classifies neuroblastic tumors into three morphologic categories: neuroblastoma, ganglioneuroblastoma, and ganglioneuroma (Fig. 66-1).
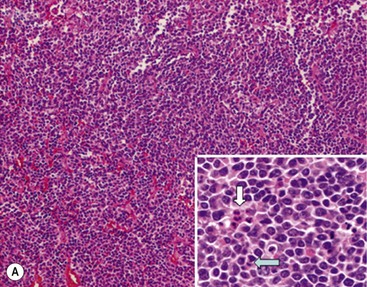
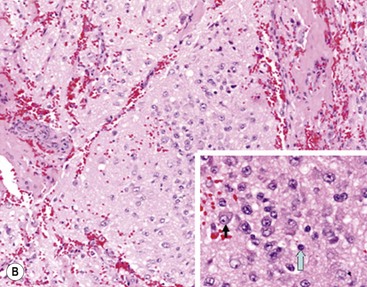
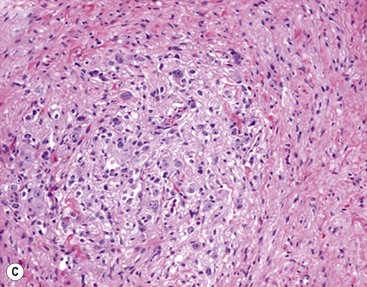
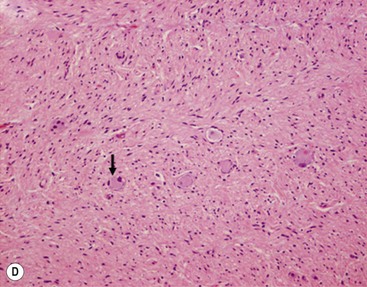
FIGURE 66-1 Histologic appearance of neuroblastic tumors. (A) An undifferentiated neuroblastoma with high MKI (10×). A clump of karyorrhectic tumor cells (white arrow) and a tumor cell undergoing mitosis (open arrow) are shown in the insert (60×). (B) A differentiating neuroblastoma, with low MKI (10×). A primitive neuroblast (gray arrow) and a differentiating tumor cell (black arrow), with features of differentiation in both the nucleus and cytoplasm, are shown in the insert (60×). Abundant neuropil is also seen. (C) A stroma-rich ganglioneuroblastoma with infrequent neuroblasts intermixed within abundant Schwannian stroma and ganglion cells (10×). (D) A stroma-rich ganglioneuroma. Ganglion cells are seen (arrow) (10×). Infiltrating lymphoid cells are also seen but no neuroblasts are present. (Courtesy of Jesse Jenkins MD and Christine Fuller MD, St. Jude Children’s Research Hospital, Memphis TN. Reprinted from Neuroblastoma, Davidoff AM. In Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott, Williams & Wilkins; 2005.)
Neuroblastomas are, by definition, Schwannian stroma poor (<50% of the tumor tissue) and can be subtyped as undifferentiated, poorly differentiated, or differentiating. Undifferentiated requires supplemental diagnostic methods such as immunohistochemistry, electron microscopy, or cytogenetics. Moreover, neuropil is not present. In poorly differentiated, <5% of tumor cells have features of differentiation, and neuropil is present. Differentiating tumors demonstrate >5% of tumor cells differentiating toward ganglion cells. To classify a cell as a differentiating neuroblast, there must be synchronous differentiation of the nucleus and eosinophilic cytoplasm.14
Additional factors that contribute to the prognostic distinction of stroma-poor neuroblastic tumors (neuroblastoma) as favorable or unfavorable subtypes include the MKI, which is defined as the number of tumor cells in mitosis or karyorrhexis per 5000 neuroblastic cells (i.e., low MKI, <100 cells; intermediate, 100–200 cells; high, >200 cells), and the patient’s age (<1.5 years, 1.5-5 years, >5 years) (Table 66-1). It has been hypothesized that neuroblastic cells with maturational potential require a latent period before demonstrating histologic evidence of differentiation. Therefore, there is a certain allowance for mitotic and karyorrhectic activities of neuroblastic cells in tumors in infants and younger children.16
TABLE 66-1
Prognostic Evaluation of Neuroblastic Tumors According to the International Neuroblastoma Pathology Classification

MKI, mitosis-karyorrhexis index.
Adapted from Shimada H, Ambros IM, Dehner LP, et al. The International NB Pathology Classification (the Shimada System). Cancer 1999;86:364–72.
The importance of this histopathologic classification was confirmed in a large, retrospective analysis reported by Shimada et al.17 The INPC classification of tumor histology provided independent prognostic information where tumors of favorable histology had a 90.8% probability of 5-year event-free survival [EFS] compared to 31.2% EFS for tumors of unfavorable histology. More recently, the INPC classification has been shown to add independent prognostic information beyond the prognostic contribution of age.18 Therefore, histopathology remains in the current multifactorial risk stratification for patients with neuroblastoma. This determination is particularly important in patients with MYCN non-amplified tumors who are older than 18 months and have stage 3 disease, or 12 to 18 months of age and have stage 4 disease, or have 4S disease. As the histopathologic pattern within a tumor can be heterogeneous, it is recommended that representative sections from at least 1 cm3 of viable, non-necrotic tissue be analyzed to determine histopathologic classification. The prognostic value of assessing the histopathology of a neuroblastoma after chemotherapy or radiation therapy has not been validated.
Stroma-rich neuroblastic tumors are classified as either ganglioneuroblastomas or ganglioneuromas. Ganglioneuroblastomas contain cells that are transitioning toward differentiation but are not completely differentiated/mature. Also, <50% of the total volume is made up of neuroblastic cells. Ganglioneuroblastomas can be further divided into ‘intermixed’ and ‘nodular’ subtypes, depending on the distribution of the neuroblastic cells. The distinction is important because of the significantly worse prognosis associated with the latter subtype, in which the neuroblastic clones that comprise grossly distinct nodules appear to be responsible for the aggressive phenotype for this subtype.16 Ganglioneuromas contain either maturing or mature cells, and lack any neuroblastomatous component. Most stroma-rich tumors (ganglioneuroblastoma, intermixed and ganglioneuroma, maturing subtype) are classified as ‘favorable’ by the INPC. However, the pathologic/prognostic classification of the ganglioneuroblastoma, nodular subtype is based on the morphologic evaluation of the neuroblastomatous nodule(s), and can, therefore, be unfavorable. Tumors that fit the criteria for ganglioneuroma, mature subtype, with abundant Schwannian stroma and fully mature ganglion cells, in the absence of neuroblasts, are considered benign, and are generally not considered for enrolment in protocols for neuroblastic tumors. Despite this, ganglioneuromas can be quite large and infiltrative, and attempts at removal can be associated with significant complications. In addition, survival does not seem to be influenced by extent of resection.19 Therefore, aggressive attempts at resection of ganglioneuromas are not recommended.
Molecular Biology
Advances in molecular biology research in the past three decades have resulted in an increased understanding of the genetic events in the pathogenesis and progression of many human malignancies, including those of childhood. Neuroblastoma, in particular, has served as a model for a molecular approach to treating patients with cancer, highlighting the utility of genetic analysis for diagnosis, risk stratification, and treatment planning. Chromosomal structural changes play a role in neuroblastoma, particularly those that result in the loss of tumor suppressors, or gain of oncogenes, gene amplification, and activating or inactivating mutations of relevant genes or their regulatory elements. The end result of alterations in these genetic elements, regardless of their specific mechanisms, is the disruption of the normal balance between cell proliferation and cell death.
DNA Content
Normal human cells contain two copies of each of 23 chromosomes; thus, a normal diploid cell has 46 chromosomes. The majority (55%) of primary neuroblastomas are triploid or ‘near-triploid/hyperdiploid’ and contain between 58 and 80 chromosomes; the remainder (45%) are either ‘near-diploid’ (35–57 chromosomes) or ‘near-tetraploid’ (81–103 chromosomes).20 The ‘DNA index’ of a tumor is the ratio of the number of chromosomes present to a diploid number of chromosome (i.e., 46). Therefore, diploid cells have a DNA index of 1.0, whereas near-triploid cells have a DNA index ranging from 1.26 to 1.76. Neuroblastomas that are near-diploid or near-tetraploid usually have structural genetic abnormalities, most frequently chromosome 1p deletion and MYCN amplification. Near-triploid or hyperdiploid tumors are characterized by almost three complete haploid sets of chromosomes with few structural abnormalities. Importantly, patients with near-triploid tumors typically have favorable clinical and biologic prognostic factors and excellent survival rates, as compared with those patients who have near-diploid or near-tetraploid tumors.21 This association is most important for infants with advanced disease as the prognostic significance of tumor ploidy appears to be lost in patients older than 2 years.22 Currently, ploidy only potentially impacts the risk group assessment of infants age 12–18 months with metastatic disease and infants with 4S disease in the COG risk stratification schema.
Amplification of MYCN
Investigation of the molecular biology of neuroblastoma began with the cytogenetic characterization of tumor-derived cell lines. These studies showed the frequent presence of extrachromosomal double-minute chromatin bodies (DMs) and chromosomally integrated homogeneously staining regions (HSRs) characteristic of gene amplification (Fig. 66-2).23 Since that time, it has been shown that the amplified region was derived from the distal short arm of chromosome 2 (2p24) and contained the MYCN proto-oncogene. MYCN encodes a 64 kDa nuclear phosphoprotein that forms a transcriptional complex by associating with other nuclear proteins expressed in the developing nervous system and other tissues.24 Enforced expression of MYCN increases the rates of DNA synthesis and cell proliferation, and shortens the G1 phase of the cell cycle.25 MYCN can also function as a classic dominant oncogene that cooperates with activated ras to transform normal cells.26 Targeted expression of MYCN in transgenic mice results in the development of neuroblastomas.27 This activity is potentiated when combined with mutations in ALK.28
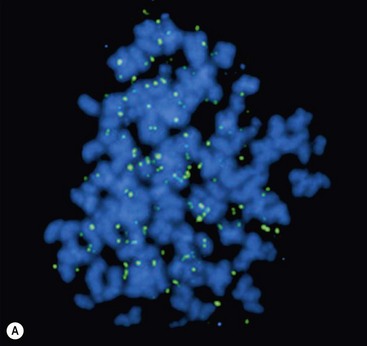
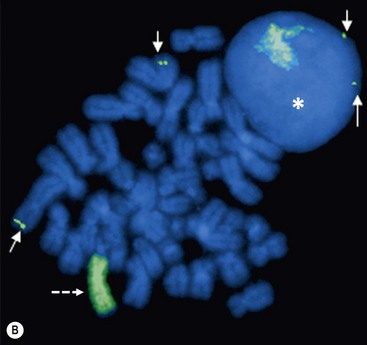
FIGURE 66-2 FISH analysis of a neuroblastoma. (A) Chromosomes in metaphase. The bright spots are double-minute chromatin bodies. (B) The metaphase chromosomes are again seen. An intact interphase nucleus is marked with an asterisk. The normal two copies of the MYCN gene are marked with solid arrows. Homogeneously staining regions (HSRs) are also seen. One is seen in the interphase nucleus, and the other is marked with a dotted arrow. (Courtesy of Marc Valentine, St. Jude Children’s Research Hospital, Memphis, TN.)
Overall, approximately 25% of primary neuroblastomas in children have MYCN amplification, with MYCN amplification being present in 40% with advanced disease but only 5–10% with low-stage disease.29 The copy number, which can range from 5- to 500-fold amplification, is usually consistent among primary and metastatic sites and at different times during tumor evolution and treatment.30 This finding suggests that MYCN amplification is an early event in the pathogenesis of neuroblastoma. Amplification of MYCN is associated with advanced stages of disease, rapid tumor progression, and poor outcome; therefore, it is a powerful prognostic indicator of tumor behavior.29,31 Amplification can be detected either by routine metaphase cytogenetics or fluorescent in situ hybridization (FISH), and current therapeutic neuroblastoma protocols have incorporated the presence or absence of MYCN amplification into their risk stratification schema.
Chromosomal Changes
Also noted on early karyotype analyses of neuroblastoma-derived cell lines were frequent deletions of the short arm of chromosome 1.32 Deletions of genetic material in tumors suggest the presence (and subsequent loss) of a tumor suppressor gene. Although no individual tumor suppressor gene has been confirmed on chromosome 1p, recent data have identified CHD5 as a strong candidate for the tumor suppressor gene that is deleted from 1p36.31 in neuroblastoma.33 Functional confirmation of the presence of a 1p tumor suppressor gene came from the demonstration that transfection of chromosome 1p into a neuroblastoma cell line resulted in morphologic changes and ultimately cell senescence.34 Approximately 20–35% of primary neuroblastomas exhibit 1p deletion, as determined by FISH, with the smallest common region of loss located within region 1p36.35 About 70% of advanced-stage neuroblastomas have 1p deletions.36 Molecular studies have shown that there is a strong correlation between 1p deletion and MYCN amplification and other high-risk features such as age older than 1 year and advanced-stage disease.35 One recent study has demonstrated that 1p deletions are independently associated with a worse outcome in patients with neuroblastoma.37
Deletion of the long arm of chromosome 11 (11q) also appears to be common in neuroblastoma, being present in about 40% of cases. Unbalanced deletion of 11q (loss with either retention or gain of 11p material) is inversely related to MYCN amplification,37,38 yet is strongly associated with other high-risk features. Recently, Attiyeh et al., on behalf of COG, showed in a large cohort of patients that unbalanced deletion of 11q and 1p36 were independently associated with a worse outcome in patients with neuroblastoma.39 Therefore, the duration of treatment for children with intermediate-risk neuroblastoma on the recent COG study was based, in part, on the 1p and 11q allelic status of the tumor.
Mutations
An example of proto-oncogene activation by point mutation involves the tyrosine kinase receptor, ALK, on the short arm of chromosome 2 (2p23). Receptor tyrosine kinases (RTK) are high-affinity cell surface receptors for many growth factors, cytokines, and hormones. When activated through ligand binding, these proteins mediate phosphorylation of tyrosine on target molecules or substrates, resulting in intracellular signaling and, ultimately, the regulation of normal cellular processes. Mutation of RTK’s can lead to constitutive activation of the signaling pathway in the absence of ligand. Recently, activating mutations of ALK have been shown to be the germline abnormality associated with hereditary neuroblastoma.40 These mutations can also be somatically acquired, as can amplification of the gene, although the prevalence of ALK activation in sporadic neuroblastoma is not known.41 Activated ALK has proven to be a targetable abnormality in neuroblastoma, with drugs such as crizotinib, an anti-ALK antibody, showing efficacy.42 Further studies have identified loss-of-function mutations in the homeobox gene PHOXB2 on 4p13 that are also associated with familial neuroblastoma, particularly when occurring together with Hirschsprung disease and/or central hypoventilation.43
Recently, inactivating mutations of ATRX, a transcriptional regulator that is part of a multiprotein complex that plays a role in regulating chromatin remodeling, nucleosome assembly, and telomere maintenance, have been found in neuroblastoma, particularly high-stage tumors in older patients.44 ATRX mutations appear to be loss-of-function mutations associated with an absence of the ATRX protein in the nucleus, and with long telomeres. How these alterations lead to lengthened telomeres is uncertain. These results may provide a molecular marker and potential therapeutic target for neuroblastoma among adolescents and young adults. It may also delineate the subset of children with neuroblastoma who have a chronic but progressive clinical course when receiving standard therapeutic approaches and who may benefit from a different treatment strategy.
Other Molecular Abnormalities
Neurotrophins and their tyrosine kinase receptors are important in the development of the sympathetic nervous system and have been implicated in the pathogenesis of neuroblastoma. Three receptor-ligand pairs have been identified: TrkA, the primary receptor for NGF; TrkB, the primary receptor of brain-derived neurotrophic factor (BDNF); and TrkC, the receptor for neurotrophin-3 (NT-3).45 TrkA appears to mediate differentiation of developing neurons or neuroblastoma in the presence of NGF ligand, and apoptosis in the absence of NGF.46 High TrkA expression is associated with favorable tumor biology and good outcome47 and is inversely correlated with MYCN amplification.48 Conversely, the TrkB/BDNF pathway appears to promote neuroblastoma survival through autocrine or paracrine signaling, especially in MYCN-amplified tumors.49 TrkB is expressed in about 40% of neuroblastomas, usually advanced-stage disease. TrkC is expressed in approximately 25% of neuroblastomas and is strongly associated with TrkA expression.50
Other molecular abnormalities frequently detected in neuroblastoma include inactivation of caspase 8, expression of CD44, and overexpression of multidrug resistance genes. Studies have demonstrated inactivation of caspase 8, a component of the Fas death-signaling complex, in MYCN-amplified neuroblastomas.51 It has been proposed that inactivation of caspase 8 renders tumor cells resistant to apoptotic signals. CD44 is a cell surface glycoprotein that appears to play a role in tumor cell adhesion.52 In neuroblastomas, CD44 expression is inversely correlated with MYCN amplification and is undetectable in most disseminated neuroblastomas.53 Multidrug resistance-associated protein (MRP) is an efflux pump whose expression in neuroblastoma appears to be correlated with MYCN amplification and poor prognosis.54,55 The presence of MRP may explain the common clinical situation in which neuroblastomas initially respond well to chemotherapy but subsequently become resistant.
Genome-Wide Association Studies
Recently, microarray technologies have generated extensive amounts of data that have aided in identifying genomic (DNA) and transcriptomic (RNA) abnormalities associated with neuroblastoma. In addition, these abnormalities have been shown to have significant predictive power when anticipating outcome for these patients.56,57 Many of these findings were generated by large scale genome-wide association studies (GWAS). This is a technique whereby all or most of the genes of patients with neuroblastoma are analyzed to find differences with the population as a whole, looking for variations that are associated with the development and aggressiveness of neuroblastoma. The causal relationship between the DNA variant associated with the cancer is uncertain but an excessive inheritance of risk variants has been postulated to increase susceptibility to the disease. Several GWAS studies have been performed in patients with neuroblastoma and have identified a number of such genetic risk variants.58 These observations suggest that developmental childhood cancers are likely influenced by common DNA variations, leading to the development of a putative genetic model.58 Recent data suggest that the higher prevalence of high-risk disease in Black and Native American patients with neuroblastoma may be associated with certain genetic variants found more commonly in these ethnic groups.59
One method for detecting CNVs is by comparative genomic hybridization (CGH). Early CGH studies showed that gain of genetic material on the long arm of chromosome 17 (17q) is perhaps the most common genetic abnormality in neuroblastomas, occurring in approximately 75% of primary tumors.60 It is unclear at this time how extra copies of 17q contribute to the malignant phenotype of neuroblastoma and which gene(s) on 17q are the critical ones. Nevertheless, gain of chromosome 17q is strongly associated with other known prognostic factors, but it may also be a powerful predictor of adverse outcome.61 More recently, GWAS studies have shown that inherited CNV at chromosome 1q21.1 is associated with neuroblastoma, implicating a neuroblastoma breakpoint family gene in early neuroblastoma genesis.62
Other studies have revealed that common genetic variation at chromosome bands 6p2263 and 2q3564 are associated with susceptibility to high-risk neuroblastoma, providing the first evidence that childhood cancers can also arise from complex interactions of polymorphic variants. More recently, a GWAS study has identified common polymorphisms including germline SNP risk alleles and somatic copy number gain, resulting in increased expression of the cysteine-rich transcriptional regulator LIM domain only 1 (LMO1) at 11p15.4. These have been shown to be strongly associated with susceptibility to developing neuroblastoma, and often are associated with advanced disease and poor survival.65
Finally, whole-genome sequencing of tumors, made possible recently by significant advances in technology, has been performed to investigate the genetic landscape of a variety of pediatric tumors.66 Initially it was felt that early alterations of genes such as MYCN may underlie the rapid acquisition of cooperating mutations in key cancer pathways through chromosome instability. However, few recurring amino acid changes have been detected in neuroblastoma specimens, suggesting that the tumor genomes were more stable than previously believed.44,67 Also, unlike the genetic landscape, the epigenetic profiles showed profound changes which suggests that epigenetic changes may have a more dominant role in pediatric tumorigenesis.
Clinical Presentation
Patients with neuroblastoma usually present with signs and symptoms that reflect the primary site and extent of disease, although localized disease is often asymptomatic. As 75% of neuroblastoma occurs in the abdominal cavity, an abdominal mass detected on physical examination is a common clinical feature, as is the complaint of abdominal pain. Other primary sites of neuroblastoma include the posterior mediastinum (20%), the cervical region (1%), and the pelvis (4%) (organ of Zuckerkandel) (Fig. 66-3). Respiratory distress or dysphagia may be a reflection of a thoracic tumor. Altered defecation or urination may be caused by mechanical compression from a pelvic tumor or by spinal cord compression by a paraspinal tumor. Spinal cord compression may also present as an altered gait. A tumor in the neck or upper thorax can produce Horner syndrome (ptosis, miosis, and anhydrosis), enophthalmos, and heterochromia of the iris. Acute cerebellar ataxia has also been observed, characterized by the dancing-eye syndrome, which includes opsoclonus, myoclonus, and chaotic nystagmus. Two-thirds of these cases occur in infants with mediastinal primary tumors.68,69 Additional signs and symptoms that reflect excessive catecholamine or vasoactive intestinal polypeptide (VIP) secretion include diarrhea, weight loss, and hypertension.
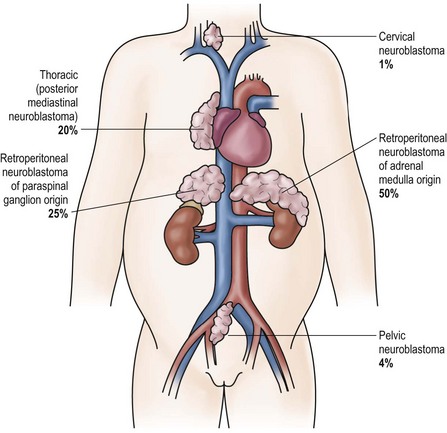
FIGURE 66-3 Primary sites for neuroblastoma are depicted in this anatomic drawing. (Reprinted from Davidoff AM. Neuroblastoma. In: Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott Williams & Wilkins; 2005.)
More than 40% of patients have metastatic disease at diagnosis. These patients are often quite ill and have systemic symptoms caused by widespread disease. Neuroblastoma in older patients has a pattern of metastatic disease in which metastases to the bone marrow, lymph nodes, and bone predominate. The frequency of involvement of distant sites is shown in Table 66-2. These metastases may manifest as bone pain from cortical metastases or anemia from marrow infiltration. The brain, spinal cord, heart, and lungs are rare sites of metastases, except with end-stage disease. Metastatic disease may be also associated with darkened eyes, referred to as ‘raccoon eyes,’ as a result of retroorbital venous plexus spread (Fig. 66-4A). This is an ominous physical sign, as is the presence of a limp in children without a history of head or extremity trauma. Infants with metastatic neuroblastoma can have stage 4S disease, which, by definition, is a localized primary tumor in patients younger than 1 year, with dissemination limited to skin, liver, or bone marrow (<10% of nucleated cells). These patients may present with ‘blueberry muffin’ cutaneous lesions (Fig. 66-4B), respiratory distress secondary to massive hepatomegaly, and anemia secondary to bone marrow disease. The diagnosis of neuroblastoma is generally made by histopathologic evaluation of primary or metastatic tumor tissue, or by the demonstration of tumor cells in the bone marrow together with elevated levels of urinary catecholamines.
TABLE 66-2
Sites of Metastases at Diagnosis for Patients with Evans Stage IV-S and Stage IV
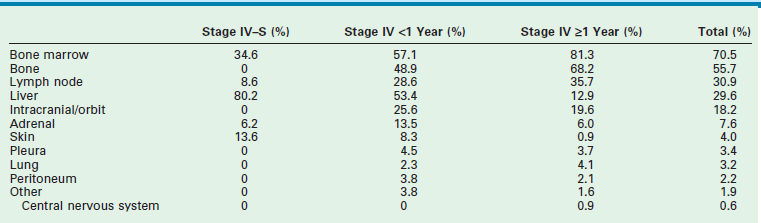
Adapted from Dubois SG, Kalika Y, Lukens JN, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. Pediatr Hematol Oncol 1999;21:181–9.
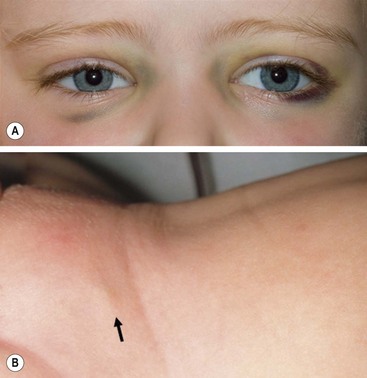
FIGURE 66-4 Clinical evidence of metastatic neuroblastoma. (A) ‘Raccoon eyes,’ characteristic of metastatic neuroblastoma in the posterior orbital venous plexus, are seen in a child with stage IV disease. (B) ‘Blueberry muffin’ spot (arrow) in the skin, characteristic of metastatic neuroblastoma, is seen in the suprapubic region of an infant with a 4S neuroblastoma. (Courtesy of Stephen Shochat, MD, St. Jude Children’s Research Hospital, Memphis, TN).
Laboratory Findings
Lactate Dehydrogenase
Despite its lack of specificity, serum lactate dehydrogenase (LDH) can have great prognostic significance. High serum levels of LDH reflect high proliferative activity or large tumor burden, and an LDH level higher than 1500 IU/L appears to be associated with a poor prognosis.70,71 Thus, LDH can be used to monitor disease activity or the response to therapy.
Ferritin
High levels of serum ferritin (>150 ng/mL) may also reflect a large tumor burden or rapid tumor progression. Elevated serum ferritin is often seen in advanced-stage neuroblastomas and indicates a poor prognosis.72 Levels often return to normal during clinical remission.
Neuron-Specific Enolase
Neuron-specific enolase (NSE) is another useful prognostic marker of advanced-stage neuroblastoma. The incidence of elevated NSE levels increases with stage.73 A serum level of NSE >100 ng/mL is associated with a poor outcome. NSE has been reported to correlate with tumor burden, suggesting its reliability as a marker of disease course.74
Catecholamine Metabolites
Neuroblastoma is characterized by the relatively unique capacity for secretion of catecholamine products, the metabolites of which can be detected in the urine of more than 90% of patients with neuroblastoma. Thus, a urine specimen is of clinical value in diagnosing neuroblastoma and determining the response to therapy. Documentation of elevated urinary catecholamines is required if the diagnosis of neuroblastoma is being made solely by the identification of neuroblasts in the bone marrow. Urinary levels of these two catabolites can also be used as markers of tumor progression or relapse, and serve as a surrogate prognostic indicator. Random urine samples are preferable to 24-hour urine estimations for younger children.75
Diagnostic Imaging
Standard Radiographs
Chest radiography can be a useful tool for demonstrating the presence of a posterior mediastinal mass, which in a child is usually a thoracic neuroblastoma. A Pediatric Oncology Group (POG) study demonstrated that a mediastinal mass was discovered on incidental chest radiographs in almost half of patients with thoracic neuroblastoma who had symptoms seemingly unrelated to their tumors.76 Abdominal radiography is less often the modality by which a neuroblastoma is discovered; however, as many as half of abdominal neuroblastomas are detectable as a mass with fine calcification.
Ultrasonography
Although ultrasonography (US) is the modality most often used during the initial assessment of a suspected abdominal mass, its sensitivity and accuracy are less than that of computed tomography (CT) or magnetic resonance imaging (MRI) for diagnosing neuoblastoma.77 These latter modalities are generally used after screening with ultrasound to assist in generating a differential diagnosis and for further anatomic definition once the presence of a mass has been confirmed.
Computed Tomography
CT can demonstrate calcification in almost 85% of neuroblastomas, and intraspinal extension of the tumor can be determined on contrast-enhanced CT.78 Overall, contrast-enhanced CT has been reported to be 82% accurate in defining neuroblastoma extent, with the accuracy increasing to nearly 97% when performed with a bone scan.79 Although some consider CT to have been supplanted by MRI, others still consider it to be the image modality of choice for patients with neuroblastoma, especially when used in conjunction with bone scintigraphy.80
Magnetic Resonance Imaging
MRI is becoming the most useful and most sensitive imaging modality for the diagnosis and staging of neuroblastoma.77,81 MRI appears to be more accurate than CT for detection of stage 4 disease. The sensitivity of MRI is 83%, and that of CT is 43%, and the specificity of MRI is 97%, and that of CT is 88%.81 Metastases to the bone and bone marrow, in particular, are better detected by MRI, as is intraspinal tumor extension (Fig. 66-5).81 When considering skeletal metastases alone, MRI and bone scan have been shown to be equivalent.81 Encasement of major vessels can be better defined by MRI than CT, especially with the use of MR angiography (see Fig. 66-5). MRI in the coronal plane is suitable for routine assessment of the whole body from the neck to the pelvis. Evaluating the utility of whole-body MRI, perhaps performed in conjunction with a functional imaging study such as positron-emission tomography (PET), is being considered for future clinical staging studies. CT and MRI are not very accurate for staging localized disease; however, the sensitivity of T1- and T2-weighted MRIs is 100% for detecting neuroblastomas in infants identified by mass screening.65
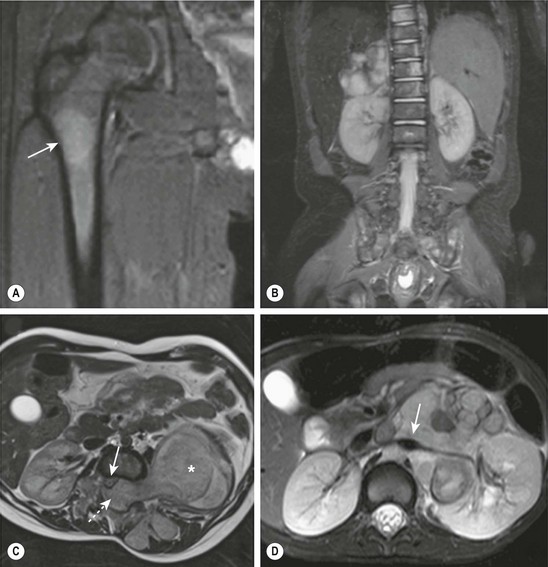
FIGURE 66-5 These MR images highlight several characteristics of high-risk neuroblastoma. (A) Bone metastasis in femur (arrow). (B) Bone marrow metastases in the vertebral bodies. (C) Intraspinal tumor extension (dotted arrow). Note displacement of spinal cord (solid arrow) from a large tumor (asterisk). (D) Encasement of major intra-abdominal vessels (arrow points to the aorta and left renal artery).
Metaiodobenzylguanidine Imaging
Metaiodobenzylguanidine (MIBG) is transported to and stored in the chromaffin cells in the same way as norepinephrine. The MIBG scintiscan is the preferred imaging study for evaluating the bone and bone marrow involvement by neuroblastoma (Fig. 66-6), having largely replaced technetium-99m methylene diphosphonate (99mTc-MDP) bone scans, which are generally inferior to MIBG in detecting skeletal or extraskeletal involvement. In addition, monitoring MDP-avid neuroblastomas by bone scintigraphy often results in false-positive imaging for months after tumor remission. Thus, 99mTc-MDP bone scanning is a second choice if MIBG imaging is not available or does not visualize known disease.82,83 Iodine-131 (131I) or iodine-123 (123I) can be used to label MIBG. 123I-MIBG supplies a reduced absorbed radiation dose and superior spatial resolution.84 The reported sensitivity of MIBG in the detection of neuroblastomas with metastases to the bone and bone marrow is 82%, and the specificity is 91%.85 Primary tumors and lymph node metastases are also detectable. MIBG can demonstrate more sites of tumor involvement in bone and bone marrow than either bone scintigraphy or standard radiography.85 However, false-negative MIBG scans have been seen in cases in which the bone scintigraphy was positive.83
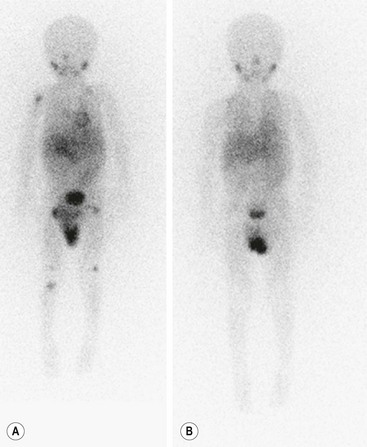
FIGURE 66-6 Imaging of neuroblastoma with MIBG scintigraphy. (A) Scan obtained at presentation of a patient with metastatic neuroblastoma. There is diffusely abnormal activity throughout much of the skeleton including the proximal right humerus, both proximal and distal femurs, and the proximal right tibia. There is also a focus of activity in the right upper retroperitoneum at the site of the primary tumor. (B) Scan obtained of the same patient after completion of therapy shows no scintigraphic evidence of MIBG-avid neuroblastoma.



