Cure Search. Children’s Oncology Group. Available at: http://www.survivorshipguidelines.org
Ries LAG et al: SEER Cancer Statistics Review, 1975–2000. Available at: http://seer.cancer.gov/csr/1975_2000
MAJOR PEDIATRIC NEOPLASTIC DISEASES
ACUTE LYMPHOBLASTIC LEUKEMIA
 General Considerations
General Considerations
Acute lymphoblastic leukemia (ALL) is the most common malignancy of childhood, accounting for about 25% of all cancer diagnoses in patients younger than age 15 years. The worldwide incidence of ALL is about 1:25,000 children per year, including 3000 children per year in the United States. The peak age at onset is 4 years; 85% of patients are diagnosed between ages 2 and 10 years. Children with Down syndrome have a 14-fold increase in the overall rate of leukemia.
ALL results from uncontrolled proliferation of immature lymphocytes. Its cause is unknown, and genetic factors may play a role. Leukemia is defined by the presence of more than 25% malignant hematopoietic cells (blasts) in the bone marrow aspirate. Leukemic blasts from the majority of cases of childhood ALL have an antigen on the cell surface called the common ALL antigen (CALLA). These blasts derive from B-cell precursors early in their development, called B-precursor ALL. Less commonly, lymphoblasts are of T-cell origin or of mature B-cell origin. Over 70% of children receiving aggressive combination chemotherapy and early presymptomatic treatment to the central nervous system (CNS) are now cured of ALL.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Presenting complaints of patients with ALL include those related to decreased bone marrow production of red blood cells (RBCs), white blood cells (WBCs), or platelets and to leukemic infiltration of extramedullary (outside bone marrow) sites. Intermittent fevers are common, as a result of either cytokines induced by the leukemia itself or infections secondary to leukopenia. Many patients present due to bruising or pallor. About 25% of patients experience bone pain, especially in the pelvis, vertebral bodies, and legs.
Physical examination at diagnosis ranges from virtually normal to highly abnormal. Signs related to bone marrow infiltration by leukemia include pallor, petechiae, and purpura. Hepatomegaly and/or splenomegaly occur in over 60% of patients. Lymphadenopathy is common, either localized or generalized to cervical, axillary, and inguinal regions. The testes may occasionally be unilaterally or bilaterally enlarged secondary to leukemic infiltration. Superior vena cava syndrome is caused by mediastinal adenopathy compressing the superior vena cava. A prominent venous pattern develops over the upper chest from collateral vein enlargement. The neck may feel full from venous engorgement. The face may appear plethoric, and the periorbital area may be edematous. A mediastinal mass can cause tachypnea, orthopnea, and respiratory distress. Leukemic infiltration of cranial nerves may cause cranial nerve palsies with mild nuchal rigidity. The optic fundi may show exudates of leukemic infiltration and hemorrhage from thrombocytopenia. Anemia can cause a flow murmur, tachycardia, and, rarely, congestive heart failure.
B. Laboratory Findings
A complete blood count (CBC) with differential is the most useful initial test because 95% of patients with ALL have a decrease in at least one cell type (single cytopenia): neutropenia, thrombocytopenia, or anemia with most patients having a decrease in at least two blood cell lines. The WBC count is low or normal (= 10,000/μL) in 50% of patients, but the differential shows neutropenia (absolute neutrophil count < 1000/μL) along with a small percentage of blasts amid normal lymphocytes. In 30% of patients the WBC count is between 10,000/μL and 50,000/μL; in 20% of patients it is over 50,000/μL, occasionally higher than 300,000/μL. Blasts are usually readily identifiable on peripheral blood smears from patients with elevated WBC counts. Peripheral blood smears also show abnormalities in RBCs, such as teardrops. Most patients with ALL have decreased platelet counts (< 150,000/μL) and decreased hemoglobin (< 11 g/dL) at diagnosis. In approximately 1% of patients diagnosed with ALL, CBCs and peripheral blood smears are entirely normal but patients have bone pain that leads to bone marrow examination. Serum chemistries, particularly uric acid and lactate dehydrogenase (LDH), are often elevated at diagnosis as a result of cell breakdown.
The diagnosis of ALL is made by bone marrow examination, which shows a homogeneous infiltration of leukemic blasts replacing normal marrow elements. The morphology of blasts on bone marrow aspirate can usually distinguish ALL from acute myeloid leukemia (AML). Lymphoblasts are typically small, with cell diameters of approximately two erythrocytes. Lymphoblasts have scant cytoplasm, usually without granules. The nucleus typically contains no nucleoli or one small, indistinct nucleolus. Immunophenotyping of ALL blasts by flow cytometry helps distinguish precursor B-cell ALL from T-cell ALL or AML. Histochemical stains specific for myeloblastic and monoblastic leukemias (myeloperoxidase and nonspecific esterase) distinguish ALL from AML. About 5% of patients present with CNS leukemia, which is defined as a cerebrospinal fluid (CSF) WBC count greater than 5/μL with blasts present on cytocentrifuged specimen.
C. Imaging
Chest radiograph may show mediastinal widening or an anterior mediastinal mass and tracheal compression secondary to lymphadenopathy or thymic infiltration, especially in T-cell ALL. Abdominal ultrasound may show kidney enlargement from leukemic infiltration or uric acid nephropathy as well as intra-abdominal adenopathy. Plain radiographs of the long bones and spine may show demineralization, periosteal elevation, growth arrest lines, or compression of vertebral bodies. Although these findings may suggest leukemia, they are not diagnostic.
 Differential Diagnosis
Differential Diagnosis
The differential diagnosis, based on the history and physical examination, includes chronic infections by Epstein-Barr virus (EBV) and cytomegalovirus (CMV), causing lymphadenopathy, hepatosplenomegaly, fevers, and anemia. Prominent petechiae and purpura suggest a diagnosis of immune thrombocytopenic purpura. Significant pallor could be caused by transient erythroblastopenia of childhood, autoimmune hemolytic anemias, or aplastic anemia. Fevers and joint pains, with or without hepatosplenomegaly and lymphadenopathy, suggest juvenile rheumatoid arthritis (JRA). The diagnosis of leukemia usually becomes straightforward once the CBC reveals multiple cytopenias and leukemic blasts. Serum LDH levels may help distinguish JRA from leukemia, as the LDH is usually normal in JRA. An elevated WBC count with lymphocytosis is typical of pertussis; however, in pertussis the lymphocytes are mature, and neutropenia is rarely associated.
 Treatment
Treatment
A. Specific Therapy
Intensity of treatment is determined by specific prognostic features present at diagnosis, the patient’s response to therapy, and specific biologic features of the leukemia cells. The majority of patients with ALL are enrolled in clinical trials designed by clinical groups and approved by the National Cancer Institute; the largest group is COG (Children’s Oncology Group). The first month of therapy consists of induction, at the end of which over 95% of patients exhibit remission on bone marrow aspirates by morphology. The drugs most commonly used in induction include oral prednisone or dexamethasone, intravenous vincristine, daunorubicin, intramuscular or intravenous asparaginase, and intrathecal methotrexate.
Consolidation is the second phase of treatment, during which intrathecal chemotherapy along with continued systemic therapy and sometimes cranial radiation therapy are given to kill lymphoblasts “hiding” in the meninges. Several months of intensive chemotherapy follows consolidation, often referred to as intensification. This intensification has led to improved survival in pediatric ALL.
Maintenance therapy can include daily oral mercaptopurine, weekly oral methotrexate, and, often, monthly pulses of intravenous vincristine and oral prednisone or dexamethasone. Intrathecal chemotherapy, either with methotrexate alone or combined with cytarabine and hydrocortisone, is usually given every 2–3 months.
Chemotherapy has significant potential side effects. Patients need to be monitored closely to prevent drug toxicities and to ensure early treatment of complications. The duration of treatment ranges between 2.2 years for girls and 3.2 years for boys in COG trials. Treatment for ALL is tailored to prognostic, or risk, groups. A child aged 1–9 years with a WBC count below 50,000/μL at diagnosis of pre B ALL and without poor biologic features [t(9;22) or an 11q23 rearrangement)] is considered to be at “standard risk” and receives less intensive therapy than a “high-risk” patient who has a WBC count at diagnosis over 50,000/μL or is 10 years of age or greater. An infant less than 1 year at diagnosis would be considered very high risk and receive even more intensive chemotherapy. Also important is the patient’s response to treatment determined by minimal residual disease (MRD) monitoring. This risk-adapted treatment approach has significantly increased the cure rate among patients with less favorable prognostic features by allowing for early intensification while minimizing treatment-related toxicities in those with favorable features. Bone marrow relapse is usually heralded by an abnormal CBC, either during treatment or following completion of therapy.
The CNS and testes are sanctuary sites of extramedullary leukemia. Currently, about one-third of all ALL relapses are isolated to these sanctuary sites. Systemic chemotherapy does not penetrate these tissues as well as it penetrates other organs. Thus, presymptomatic intrathecal chemotherapy is a critical part of ALL treatment, without which many more relapses would occur in the CNS, with or without bone marrow relapse. The majority of isolated CNS relapses are diagnosed in an asymptomatic child at the time of routine intrathecal injection, when CSF cell count and differential shows an elevated WBC with leukemic blasts. Occasionally, symptoms of CNS relapse develop: headache, nausea and vomiting, irritability, nuchal rigidity, photophobia, changes in vision, and cranial nerve palsies. Currently, testicular relapse occurs in less than 5% of boys. The presentation of testicular relapse is usually unilateral painless testicular enlargement, without a distinct mass. Routine follow-up of boys both on and off treatment includes physical examination of the testes.
Bone marrow transplantation, now called hematopoietic stem cell transplantation (HSCT), is rarely used as initial treatment for ALL, because most patients are cured with chemotherapy alone. Patients whose blasts contain certain chromosomal abnormalities, such as t(9;22) or hypodiploidy (< 44 chromosomes), and patients with a very slow response to therapy may have a better cure rate with early HSCT from a human leukocyte antigen (HLA)-DR–matched sibling donor, or perhaps a matched unrelated donor, than with intensive chemotherapy alone. HSCT cures about 50% of patients who relapse, provided that a second remission is achieved with chemotherapy before transplant. Children who relapse more than 1 year after completion of chemotherapy (late relapse) may be cured with intensive chemotherapy without HSCT.
Several new biologic agents, including tyrosine kinase inhibitors and immunotoxins, are currently in various stages of research, development, and in chemotherapeutic trials. Some of these therapies may prove relevant for future treatment of poor risk or relapsed ALL.
Several years ago, Imatinib, a tyrosine kinase inhibitor (TKI), directed against the Philadelphia chromosome (Ph+) protein product, was combined in a backbone of intensive ALL therapy for Ph+ ALL in pediatric patients. The preliminary results of this trial showed a 3-year event-free survival (EFS) of 78% compared to about 50% in historical controls. An ongoing trial for COG in Ph+ ALL is incorporating a newer, more targeted TKI, Dasatinib, into a very similar intensive chemotherapy background with the goal of improving EFS further for this select group of patients. As more is understood about the biology of ALL, further therapy will likely include more of these targeted agents.
B. Supportive Care
Tumor lysis syndrome, which consists of hyperkalemia, hyperuricemia, hyperphosphatemia, should be anticipated when treatment is started. Maintaining brisk urine output with intravenous fluids plus/minus alkalinization of urine with intravenous sodium bicarbonate, and treating with oral allopurinol are appropriate steps in managing tumor lysis syndrome. Rasburicase is indicated for severe tumor lysis syndrome with initial high uric acid values or high WBC at presentation. Serum levels of potassium, phosphorus, and uric acid should be monitored. If superior vena caval or superior mediastinal syndrome is present, general anesthesia is contraindicated temporarily and until there has been some decrease in the mass. If hyperleukocytosis (WBC count > 100,000/μL) is accompanied by hyperviscosity with symptoms of respiratory distress and/or mental status changes, leukophoresis may be indicated to rapidly reduce the number of circulating blasts and minimize the potential thrombotic or hemorrhagic CNS complications. Throughout the course of treatment, all transfused blood and platelet products should be irradiated to prevent graft-versus-host disease from the transfused lymphocytes. Whenever possible, blood products should be leukodepleted to minimize CMV transmission, transfusion reactions, and sensitization to platelets.
Due to the immunocompromised state of the patient with ALL, bacterial, fungal, and viral infections are serious and can be life-threatening or fatal. During the course of treatment, fever (temperature = 38.3°C) and neutropenia (absolute neutrophil count < 500/μL) require prompt assessment, blood cultures from each lumen of a central line, and prompt treatment with empiric broad-spectrum antibiotics. Patients receiving ALL treatment must receive prophylaxis against Pneumocystis jiroveci (formerly Pneumocystis carinii). Trimethoprim–sulfamethoxazole given twice each day on 2 or 3 consecutive days per week is the drug of choice. Patients who are nonimmune to varicella are at risk for very serious—even fatal—infection. Such patients should receive varicella-zoster immune globulin (VZIG) within 72 hours after exposure and treatment with intravenous acyclovir for active infection.
 Prognosis
Prognosis
Cure rates depend on specific prognostic features present at diagnosis, biologic features of the leukemic blast, and the response to therapy. Two of the most important features are WBC count and age. Children aged 1–9 years whose diagnostic WBC count is less than 50,000/μL, standard risk ALL, have an EFS in the 90% range, while children 10 years of age or older have an EFS of approximately 88%. Minimal residual disease (MRD) measurements are now frequently used to determine both the rapidity of response as well as the depth of remission attained at the end of induction (first 4–6 weeks of therapy). Patients with very low levels of MRD at the end of induction will likely have a superior EFS as compared to other patients with similar initial risk factors but a higher MRD level. On the flip side, by identifying patients with higher MRD at end induction, more intensified therapy can be delivered in order to reduce the MRD and thus, improve the EFS.
Certain chromosomal abnormalities present in the leukemic blasts at diagnosis influence prognosis. Patients with t(9;22), the Philadelphia chromosome, had a poor chance of cure in the past but as discussed earlier in this chapter, now have improved outcome with the incorporation of a directed TKI. Likewise, infants younger than age 6 months with t(4;11) have a poor chance of cure with conventional chemotherapy. In contrast, patients whose blasts are hyperdiploid (containing > 50 chromosomes instead of the normal 46) with trisomies of chromosomes 4, and 10, and patients whose blasts have a t(12;21) and ETV6-AML1 rearrangement have a greater chance of cure, approaching 95%–97% EFS, than do children without these characteristics.
Advani AS, Hunger SP, Burnett AK: Acute leukemia in adolescents and young adults. Semin Oncol 2009;36(3):213 [PMID: 19460579].
Gaynon PS et al: Long term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a children’s oncology group report. Leukemia 2010:24(2):285 [PMID: 20016531].
Hochberg J, Khaled S, Forman SJ, Cairo MS: Criteria for and outcomes of allogeneic haematopoietic stem cell transplant in children, adolescents and young adults with acute lymphoblastic leukaemia in first complete remission. Br J Haematol 2013;161(1):27 [PMID: 23384118].
Martin A, Morgan E, Hijiya N: Relapsed or refractory pediatric acute lymphoblastic leukemia: current and emerging treatments. Paediatr Drugs 2012;14(6):377 [PMID: 22880941].
Nguyen K et al: Factors influencing survival after relapse from acute lymphoblastic leukemia: a children’s oncology group study. Leukemia 2008;22(12):2142 [PMID: 18818707].
Schultz KR et al: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. JCO 2009;27(31): 5175 [PMID: 19805687].
Vrooman LM, Silverman LB: Childhood acute lymphoblastic leukemia: update on prognostic factors. Curr Opin Pediatr 2009;21(1):1 [PMID: 19242236].
ACUTE MYELOID LEUKEMIA
 General Considerations
General Considerations
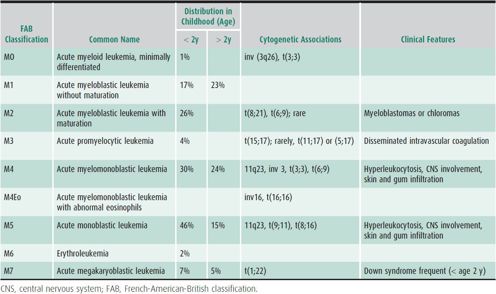
Approximately 500 new cases of AML occur per year in children and adolescents in the United States. Although AML accounts for only 25% of all leukemias in this age group, it is responsible for at least one-third of deaths from leukemia in children and teenagers. Congenital conditions associated with an increased risk of AML include Diamond-Blackfan anemia; neurofibromatosis; Down syndrome; Wiskott-Aldrich, Kostmann, and Li-Fraumeni syndromes; as well as chromosomal instability syndromes such as Fanconi anemia. Acquired risk factors include exposure to ionizing radiation, cytotoxic chemotherapeutic agents, and benzenes. However, the vast majority of patients have no identifiable risk factors. Historically, the diagnosis of AML was based almost exclusively on morphology and immunohistochemical staining of the leukemic cells. AML has eight subtypes (M0–M7) according to the French-American-British (FAB) classification (Table 31–1). Immunophenotypic, cytogenetic, and molecular analyses are increasingly important in confirming the diagnosis of AML and subclassifying it into biologically distinct subtypes that have therapeutic and prognostic implications. Recently the World Health Organization (WHO) classification was published to describe AML as AML with recurrent genetic abnormalities with a list of genetic abnormalities sufficient to diagnose AML and then AML not otherwise specified with morphologic descriptions of AML including similar to the FAB classification. Cytogenetic clonal abnormalities occur in 80% of patients with AML and are often predictive of outcome.
Table 31–1. FAB subtypes of acute myeloid leukemia.
Aggressive induction therapy currently results in a 75%–85% complete remission rate. However, long-term survival has improved only modestly to approximately 50%, despite the availability of several effective agents, improvements in supportive care, and increasingly intensive therapies.
 Clinical Findings
Clinical Findings
The clinical manifestations of AML commonly include anemia (44%), thrombocytopenia (33%), and neutropenia (69%). Symptoms may be few and innocuous or may be life-threatening. The median hemoglobin value at diagnosis is 7 g/dL, and platelets usually number fewer than 50,000/μL. Frequently the absolute neutrophil count is under 1000/μL, although the total WBC count is over 100,000/μL in 25% of patients at diagnosis.
Hyperleukocytosis may be associated with life-threatening complications. Venous stasis and sludging of blasts in small vessels cause hypoxia, hemorrhage, and infarction, most notably in the lung and CNS. This clinical picture is a medical emergency requiring rapid intervention, such as leukophoresis, to decrease the leukocyte count. CNS leukemia is present in 5%–15% of patients at diagnosis, a higher rate of initial involvement than in ALL. Certain subtypes, such as M4 and M5, have a higher likelihood of meningeal infiltration than do other subtypes. Additionally, clinically significant coagulopathy may be present at diagnosis in patients with M3, M4, or M5 subtypes. This problem manifests as bleeding or an abnormal disseminated intravascular coagulation screen and should be at least partially corrected prior to initiation of treatment, which may transiently exacerbate the coagulopathy.
 Treatment
Treatment
A. Specific Therapy
AML is less responsive to treatment than ALL and requires more intensive chemotherapy. Toxicities from therapy are common and likely to be life-threatening; therefore, treatment should be undertaken only at a tertiary pediatric oncology center.
Current AML protocols rely on intensive administration of anthracyclines, cytarabine, and etoposide for induction of remission. After remission is obtained, patients who have a matched sibling donor undergo allogeneic HSCT, while those without an appropriate related donor are treated with additional cycles of aggressive chemotherapy for a total of 4–5 cycles. Inv16 and t(8;21) herald a more responsive subtype of AML. In patients with a rapid response to induction chemotherapy, intensive chemotherapy alone may be curative in patients whose blasts harbor these cytogenetic abnormalities. Additional recognized genetic risk factors that carry a poor outcome for children with AML include monosomy 7 and FLT3 internal tandem duplications (ITD). HSCT is recommended for all these patients, using either a related or unrelated donor. Trials with risk grouping are ongoing as more is understood about the varying biologic factors.
The biologic heterogeneity of AML is becoming increasingly important therapeutically. The M3 subtype, associated with t(15;17) demonstrated either cytogenetically or molecularly, is currently treated with all trans-retinoic acid in addition to chemotherapy with high-dose cytarabine and daunorubicin. All trans-retinoic acid leads to differentiation of promyelocytic leukemia cells and can induce remission, but cure requires conventional chemotherapy as well. The use of arsenic trioxide has also been investigated in the treatment of this subtype of AML with favorable results. This subtype has an increased event-free survival over other AML subtypes.
Another biologically distinct subtype of AML occurs in children with Down syndrome, M7, or megakaryocytic AML. Using less intensive treatment, remission induction rate and overall survival of these children are dramatically superior to non–Down syndrome children with AML. It is important that children with Down syndrome receive appropriate treatment specifically designed to be less intensive due to their increased rate of toxicity with chemotherapeutic agents.
As with ALL, newer biologic agents with more specific targeting are available and undergoing clinical trails. One such agent, sorafenib, appears to be active against AML with Flt3 ITDs. Combining sorafenib with AML therapy has been useful in relapsed disease and is now being studied in to upfront trials.
Clofarabine, a newer nucleoside analogue, also has activity in AML and is currently undergoing trials in relapsed and refractory patients with promising results.
B. Supportive Care
Tumor lysis syndrome rarely occurs during induction treatment of AML. Nevertheless, when the diagnostic WBC cell count is greater than 100,000/μL or significant adenopathy or organomegaly is present, one should maintain brisk urine output, and follow potassium, uric acid, and phosphorous laboratory values closely. Hyperleukocytosis (WBC > 100,000/μL) is a medical emergency and, in a symptomatic patient, requires rapid intervention such as leukophoresis to rapidly decrease the number of circulating blasts and thereby decrease hyperviscosity. Delaying transfusion of packed RBCs until the WBC can be decreased to below 100,000/μL avoids exacerbating hyperviscosity. It is also important to correct the coagulopathy commonly associated with M3, M4, or M5 subtypes prior to beginning induction chemotherapy. As with the treatment of ALL, all blood products should be irradiated and leukodepleted; Pneumocystis prophylaxis must be administered during treatment and for several weeks afterward; and patients not immune to varicella must receive VZIG within 72 hours of exposure and prompt treatment with intravenous acyclovir for active infection.
Onset of fever (temperature ≥ 38.3°C) or chills associated with neutropenia requires prompt assessment, blood cultures from each lumen of a central venous line, other cultures such as throat or urine as appropriate and prompt initiation of broad-spectrum intravenous antibiotics. Infections in this population of patients can rapidly become life-threatening. Because of the high incidence of invasive fungal infections, there should be a low threshold for initiating antifungal therapy. Filgrastim (granulocyte colony-stimulating factor) may be used to stimulate granulocyte recovery during the treatment of AML and results in shorter periods of neutropenia and hospitalization. It must be stressed that the supportive care for this group of patients is as important as the leukemia-directed therapy and that this treatment should be carried out only at a tertiary pediatric cancer center.
 Prognosis
Prognosis
Published results from various centers show a 50%–60% survival rate at 5 years following first remission for patients who do not have matched sibling hematopoietic stem cell donors. Patients with matched sibling donors fare slightly better, with 5-year survival rates of 60%–70% after allogeneic HSCT.
As treatment becomes more sophisticated, outcome is increasingly related to the subtype of AML. Currently, AML in patients with t(8;21), t(15;17), inv 16, or Down syndrome has the most favorable prognosis, with 65%–75% long-term survival using modern treatments, including chemotherapy alone. The least favorable outcome occurs in AML patients with monosomy 7 or 5, 7q, 5q–, 11q23 cytogenetic abnormalities, or FLT 3 mutations or ITD.
Gibson BE, Webb DK, Howman AJ, De Graaf SS, Harrison CJ, Wheatley K: Results of a randomized trial in children with acute myeloid leukaemia: Medical Research Council AML12 trial. Br J Haematol 2011 Nov;155(3):366–376 [PMID: 21902686].
Meshinchi S, Arceci RJ: Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist 2007;12:341 [PMID: 17405900].
Neudorf S et al: Allogeneic bone marrow transplantation for children with acute myelocytic leukemia in first remission demonstrates a role for graft versus leukemia in the maintenance of disease-free survival. Blood 2004;103:3655 [PMID: 14751924].
Rubnitz JE, Inaba H: Childhood acute myeloid leukaemia. Br J Haematol 2012;159(3):259 [PMID: 22966788].
Sternherz PG et al: Clofarabine induced durable complete remission in heavily pretreated adolescents with relapsed and refractory leukemia. J Pediatr Hematol Oncol 2007;29:656 [PMID: 17805046].
Vardiman J et al: The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937.
MYELOPROLIFERATIVE DISEASES
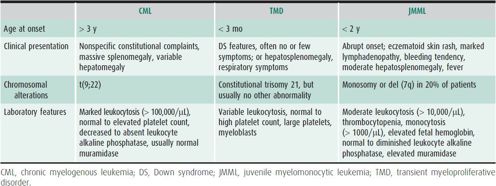
Myeloproliferative diseases in children are relatively rare. They are characterized by ineffective hematopoiesis that results in excessive peripheral blood counts. The three most important types are chronic myelogenous leukemia (CML), which accounts for less than 5% of the childhood leukemias, transient myeloproliferative disorder in children with Down syndrome, and juvenile myelomonocytic leukemia (Table 31–2).
Table 31–2. Comparison of JMML, CML, and TMD.
1. Chronic Myelogenous Leukemia
 General Considerations
General Considerations
CML with translocation of chromosomes 9 and 22 (the Philadelphia chromosome, Ph+) is identical to adult Ph+CML. Translocation 9;22 results in the fusion of the BCR gene on chromosome 22 and the ABL gene on chromosome 9. The resulting fusion protein is a constitutively active tyrosine kinase that interacts with a variety of effector proteins and allows for deregulated cellular proliferation, decreased adherence of cells to the bone marrow extracellular matrix, and resistance to apoptosis. The disease usually progresses within 3 years to an accelerated phase and then to a blast crisis. It is generally accepted that Ph+ cells have an increased susceptibility to the acquisition of additional molecular changes that lead to the accelerated and blast phases of disease.
 Clinical Findings
Clinical Findings
Patients with CML may present with nonspecific complaints similar to those of acute leukemia, including bone pain, fever, night sweats, and fatigue. However, patients can also be asymptomatic. Patients with a total WBC count of more than 100,000/μL may have symptoms of leukostasis, such as dyspnea, priapism, or neurologic abnormalities. Physical findings may include fever, pallor, ecchymoses, and hepatosplenomegaly. Anemia, thrombocytosis, and leukocytosis are frequent laboratory findings. The peripheral smear is usually diagnostic, with a characteristic predominance of myeloid cells in all stages of maturation, increased basophils and relatively few blasts.
 Treatment & Prognosis
Treatment & Prognosis
Historically, hydroxyurea or busulfan has been used to reduce or eliminate Ph+ cells and HSCT was the only consistently curative intervention. Reported survival rates for patients younger than age 20 years transplanted in the chronic phase from matched-related donors are 70%–80%. Unrelated stem cell transplants result in survival rates of 50%–65%.
The understanding of the molecular mechanisms involved in the pathogenesis of CML has led to the rational design of molecularly targeted therapy. Imatinib mesylate (Gleevec) is a tyrosine kinase inhibitor that has had dramatic success in the treatment of CML, with most adults and children achieving cytogenetic remission. There are now newer, more targeted TKIs including dasatinib, erlotinib, nilotinib, and ponatinib. These medications in adults have an increased incidence of molecular remissions and may be all that is required for long-term survival in adults. The durability of the remission for children with TKIs therapy alone is unclear but is now the accepted upfront therapy.
2. Transient Myeloproliferative Disorder
Transient myeloproliferative disorder is unique to patients with trisomy 21 or mosaicism for trisomy 21. It is characterized by uncontrolled proliferation of blasts, usually of megakaryocytic origin, during early infancy and spontaneous resolution. The pathogenesis of this process is not well understood, although mutations in the GATA1 gene have recently been implicated as initial events.
Although the true incidence is unknown, it is estimated to occur in up to 10% of patients with Down syndrome. Despite the fact that the process usually resolves by 3 months of age, organ infiltration may cause significant morbidity and mortality.
Patients can present with hydrops fetalis, pericardial or pleural effusions, or hepatic fibrosis. More frequently, they are asymptomatic or only minimally ill. Therefore, treatment is primarily supportive. Patients without symptoms are not treated, and those with organ dysfunction receive low doses of chemotherapy or leukophoresis (or both) to reduce peripheral blood blast counts. Although patients with transient myeloproliferative disorder have apparent resolution of the process, approximately 30% go on to develop acute megakaryoblastic leukemia (AML M7) within 3 years.
3. Juvenile Myelomonocytic Leukemia
Juvenile myelomonocytic leukemia (JMML) accounts for approximately one-third of the myelodysplastic and myeloproliferative disorders in childhood. Patients with neurofibromatosis type 1 (NF-1) are at higher risk of JMML than the general population. It typically occurs in infants and very young children and is occasionally associated with monosomy 7 or a deletion of the long arm of chromosome 7.
Patients with JMML present similarly to those with other hematopoietic malignancies, with lymphadenopathy, hepatosplenomegaly, skin rash, or respiratory symptoms. Patients may have stigmata of NF-1 with neurofibromas or café au lait spots. Laboratory findings include anemia, thrombocytopenia, leukocytosis with monocytosis, and elevated fetal hemoglobin.
The results of chemotherapy for children with JMML have been disappointing, with estimated survival rates of less than 30%. Approximately 40%–45% of patients are projected to survive long term using HSCT, although optimizing conditioning regimens and donor selection may improve these results.
Crispino JD: GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr Blood Cancer 2005;44:40 [PMID: 15390312].
Gassas A et al: A basic classification and a comprehensive examination of pediatric myeloproliferative syndromes. J Pediatr Hematol Oncol 2005;27:192 [PMID: 15838389].
Lee SJ et al: Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood 2008;112(8):3500 [PMID: 18664621].
Mundschau G et al: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 2003;101:4298 [PMID: 12560215].
Passmore SJ et al: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 2003;121:758 [PMID: 12780790].
Pulsipher MA: Treatment of CML in pediatric patients: should imatinib mesylate (STI-571, Gleevec) or allogeneic hematopoietic cell transplant be front-line therapy? Pediatr Blood Cancer 2004;43:523 [PMID: 15382266].
Snyder DS: New approved dasatinib regimen available for clinical use. Expert Rev Anticancer Ther 2009;9(3):285 [PMID: 19275507].
Suttorp M: Innovative approaches of targeted therapy for CML of childhood in combination with paediatric haematopoietic SCT. Bone Marrow Transplant 2008;42(Suppl 2):S40 [PMID: 18978743].
Vardiman JW: Chronic myelogenous leukemia, BCR-ABL1+. Am J Clin Pathol 2009;132(2):250 [PMID: 19605820].
BRAIN TUMORS
 General Considerations
General Considerations
The classic triad of morning headache, vomiting, and papilledema is present in fewer than 30% of children at presentation. School failure and personality changes are common in older children. Irritability, failure to thrive, and delayed development are common in very young children with brain tumors. Recent-onset head tilt can result from a posterior fossa tumor.
Brain tumors are the most common solid tumors of childhood, accounting for 1500–2000 new malignancies in children each year in the United States and for 25%–30% of all childhood cancers. In general, children with brain tumors have a better prognosis than do adults. Favorable outcome occurs most commonly with low-grade and fully resectable tumors. Unfortunately, cranial irradiation in young children can have significant neuropsychological, intellectual, and endocrinologic sequelae.
Brain tumors in childhood are biologically and histologically heterogeneous, ranging from low-grade localized lesions to high-grade tumors with neuraxis dissemination. High-dose systemic chemotherapy is used frequently, especially in young children with high-grade tumors, in an effort to delay, decrease, or completely avoid cranial irradiation. Such intensive treatment may be accompanied by autologous HSCT or peripheral stem cell reconstitution.
The causes of most pediatric brain tumors are unknown. The risk of developing astrocytomas is increased in children with neurofibromatosis or tuberous sclerosis. Several studies show that some childhood brain tumors occur in families with increased genetic susceptibility to childhood cancers in general, brain tumors, or leukemia and lymphoma. A higher incidence of seizures has been observed in relatives of children with astrocytoma. The risk of developing a brain tumor is increased in children who received cranial irradiation for treatment of meningeal leukemia. All children with gliomas and meningiomas should be screened for NF-1. In children with meningiomas, without the skin findings of NF-1, neurofibromatosis type 2 and Von Hippel-Lindau syndrome should be considered. Inherited germline mutations are possible in atypical teratoid/rhabdoid tumors (AT/RTs) and in choroid plexus carcinomas. Careful family histories should be taken in these tumors and genetic counseling considered.
Because pediatric brain tumors are rare, they are often misdiagnosed or diagnosed late; most pediatricians see no more than two children with brain tumors during their careers.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Clinical findings at presentation vary depending on the child’s age and the tumor’s location. Children younger than age 2 years more commonly have infratentorial tumors. Children with such tumors usually present with nonspecific symptoms such as vomiting, unsteadiness, lethargy, and irritability. Signs may be surprisingly few or may include macrocephaly, ataxia, hyperreflexia, and cranial nerve palsies. Because the head can expand in young children, papilledema is often absent. Measuring head circumference and observing gait are essential in evaluating a child for possible brain tumor. Eye findings and apparent visual disturbances such as difficulty tracking can occur in association with optic pathway tumors such as optic glioma. Optic glioma occurring in a young child is often associated with neurofibromatosis.
Older children more commonly have supratentorial tumors, which are associated with headache, visual symptoms, seizures, and focal neurologic deficits. Initial presenting features are often nonspecific. School failure and personality changes are common. Vaguely described visual disturbance is often present, but the child must be directly asked. Headaches are common, but they often will not be predominantly in the morning. The headaches may be confused with migraine.
Older children with infratentorial tumors characteristically present with symptoms and signs of hydrocephalus, which include progressively worsening morning headache and vomiting, gait unsteadiness, double vision, and papilledema. Cerebellar astrocytomas enlarge slowly, and symptoms may worsen over several months. Morning vomiting may be the only symptom of posterior fossa ependymomas, which originate in the floor of the fourth ventricle near the vomiting center. Children with brainstem tumors may present with facial and extraocular muscle palsies, ataxia, and hemiparesis; hydrocephalus occurs in approximately 25% of these patients at diagnosis.
B. Imaging and Staging
In addition to the tumor biopsy, neuraxis imaging studies are obtained to determine whether dissemination has occurred. It is unusual for brain tumors in children and adolescents to disseminate outside the CNS.
Magnetic resonance imaging (MRI) has become the preferred diagnostic study for pediatric brain tumors. MRI provides better definition of the tumor and delineates indolent gliomas that may not be seen on computed tomography (CT) scan. In contrast, a CT scan can be done in less than 10 minutes—as opposed to the 30 minutes required for an MRI scan—and is still useful if an urgent diagnostic study is necessary or to detect calcification of a tumor. Both scans are generally done with and without contrast enhancement. Contrast enhances regions where the blood-brain barrier is disrupted. Postoperative scans to document the extent of tumor resection should be obtained within 48 hours after surgery to avoid postsurgical enhancement.
Imaging of the entire neuraxis and CSF cytologic examination should be part of the diagnostic evaluation for patients with tumors such as medulloblastoma, ependymoma, and pineal region tumors. Diagnosis of neuraxis drop metastases (tumor spread along the neuraxis) can be accomplished by gadolinium-enhanced MRI incorporating sagittal and axial views. MRI of the spine should be obtained preoperatively in all children with midline tumors of the fourth ventricle or cerebellum. A CSF sample should be obtained during the diagnostic surgery or, if that is not possible, 7 to 10 days after the surgery. Lumbar CSF is preferred over ventricular CSF for cytologic examination. Levels of biomarkers in the blood and CSF, such as human chorionic gonadotropin and α-fetoprotein, may be helpful at diagnosis and in follow-up. Both human chorionic gonadotropin and α-fetoprotein should be obtained from the blood preoperatively for all pineal and suprasellar tumors.
The neurosurgeon should discuss staging and sample collection with an oncologist before surgery in a child newly presenting with a scan suggestive of brain tumor.
C. Classification
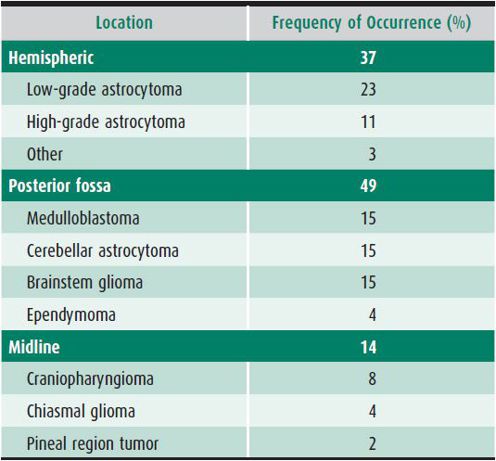
About 50% of the common pediatric brain tumors occur above the tentorium and 50% in the posterior fossa. In the very young child, posterior fossa tumors are more common. Most childhood brain tumors can be divided into two categories according to the cell of origin: (1) glial tumors, such as astrocytomas and ependymomas, or (2) nonglial tumors, such as medulloblastoma and other primitive neuroectodermal tumors. Some tumors contain both glial and neural elements (eg, ganglioglioma). A group of less common CNS tumors does not fit into either category (ie, craniopharyngiomas, AT/RTs, germ cell tumors, choroid plexus tumors, and meningiomas). Low- and high-grade tumors are found in most categories. Table 31–3 lists the locations and frequencies of the common pediatric brain tumors.
Table 31–3. Location and frequency of common pediatric brain tumors.
Astrocytoma is the most common brain tumor of childhood. Most are juvenile pilocytic astrocytoma (WHO grade I) found in the posterior fossa with a bland cellular morphology and few or no mitotic figures. Low-grade astrocytomas are in many cases curable by complete surgical excision alone. Chemotherapy may be effective in about 40%–50% of low-grade astrocytomas and even in those who will fail chemotherapy eventually, it may delay time to radiation.
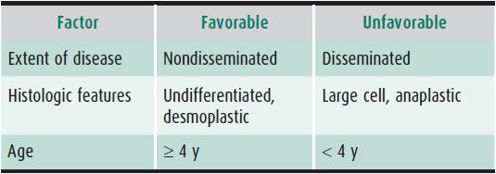
Medulloblastoma and related primitive neuroectodermal tumors are the most common high-grade brain tumors in children. These tumors usually occur in the first decade of life, with a peak incidence between ages 5 and 10 years and a female-male ratio of 2.1:1.3. The tumors typically arise in the midline cerebellar vermis, with variable extension into the fourth ventricle. Neuraxis dissemination at diagnosis affects from 10% to 46% of patients. Prognostic factors are outlined in Table 31–4. Determination of risk using histology may soon be replaced by molecular classifications.
Table 31–4. Prognostic factors in children with medulloblastoma.
Brainstem tumors are third in frequency of occurrence in children. They are frequently of astrocytic origin and often are high grade. Children with tumors that diffusely infiltrate the brainstem and involve primarily the pons (diffuse intrinsic pontine gliomas) have a long-term survival rate of less than 5%. There has been considerable biological discovery, largely from autopsy samples, in diffuse pontine gliomas in the very recent past; it is hoped this will result in new, better targeted therapies. Brainstem tumors that occur above or below the pons and grow in an eccentric or cystic manner have a somewhat better outcome. Exophytic tumors in this location may be amenable to surgery. Generally, brainstem tumors are treated without a tissue diagnosis.
Other brain tumors such as ependymomas, germ cell tumors, choroid plexus tumors, and craniopharyngiomas are less common, and each is associated with unique diagnostic and therapeutic challenges.
 Treatment
Treatment
A. Supportive Care
Dexamethasone should be started prior to initial surgery (0.5–1.0 mg/kg initially, then 0.25–0.5 mg/kg/d in four divided doses). Anticonvulsants should be started if the child has had a seizure or if the surgical approach is likely to induce seizures. Keppra is now the preferred anticonvulsant in this population as it does not induce liver enzymes. Because postoperative treatment of young children with high-grade brain tumors incorporates increasingly more intensive systemic chemotherapy, consideration should also be given to the use of prophylaxis for prevention of oral candidiasis and Pneumocystis infection. Dexamethasone potentially reduces the effectiveness of chemotherapy and should be discontinued as soon after surgery as possible.
Optimum care for the pediatric patient with a brain tumor requires a multidisciplinary team including subspecialists in pediatric neurosurgery, neuro-oncology, neurology, endocrinology, neuropsychology, radiation therapy, and rehabilitation medicine, as well as highly specialized nurses, social workers, and staff in physical therapy, occupational therapy, and speech and language science.
B. Specific Therapy
The goal of treatment is to eradicate the tumor with the least short- and long-term morbidity. Long-term neuropsychological morbidity becomes an especially important issue related to deficits caused by the tumor itself and the sequelae of treatment. Meticulous surgical removal of as much tumor as possible is generally the preferred initial approach. Technologic advances in the operating microscope, the ultrasonic tissue aspirator, and the CO2 laser (which is less commonly used in pediatric brain tumor surgery); the accuracy of computerized stereotactic resection; and the availability of intraoperative monitoring techniques such as evoked potentials and electrocorticography have increased the feasibility and safety of surgical resection of many pediatric brain tumors. Second-look surgery after chemotherapy is increasingly being used when tumors are incompletely resected at initial surgery.
Radiation therapy for pediatric brain tumors is in a state of evolution. For tumors with a high probability of neuraxis dissemination (eg, medulloblastoma), craniospinal irradiation is still standard therapy in children older than age 3 years. For tumors, such as medulloblastomas, there are ongoing trials aimed at further reducing the craniospinal dose. In others (eg, ependymoma), craniospinal irradiation has been abandoned because neuraxis dissemination at first relapse is rare. Conformal radiation and the use of three-dimensional treatment planning are now in routine.
Chemotherapy is effective in treating low-grade and malignant astrocytomas and medulloblastomas. There is an initial report supporting the effectiveness of chemotherapy in AT/RTs. The therapy used in this study was prolonged and intensive. The utility of chemotherapy in ependymoma is being re-explored in national trials. A series of brain tumor protocols for children younger than age 3 years involved administering intensive chemotherapy after tumor resection and delaying or omitting radiation therapy. The results of these trials have generally been disappointing but have taught valuable lessons regarding the varying responses to chemotherapy of different tumor types. Superior results seem to have been obtained in the very young with high-dose chemotherapy strategies with stem cell rescue often followed by conformal radiotherapy. Conformal techniques allow the delivery of radiation to strictly defined fields and may limit side effects.
Perhaps the most exciting development in pediatric neuro-oncology is the development of biologically and clinically relevant subclassifications in both medulloblastoma and ependymoma. This development will drive a new generation of targeted therapy aimed at these biologically defined groups. The consensus definition of four biologically defined entities in medulloblastoma, including the Wnt and SHH groups, is the best example of this. New studies are in the planning stage based on this new defined biology.
In older children with malignant glioma, the current approach is surgical resection of the tumor and combined-modality treatment with irradiation and intensive chemotherapy. It has recently been realized there is considerable heterogeneity in pediatric high-grade gliomas. Some, such as the congenital tumors, may do well with relatively modest therapy. Others, such as epithelioid glioblastomas may harbor Braf mutations and may be targetable with specific agents. Generally, however, the prognosis is poor for children with high-grade gliomas and there has been little progress in finding better chemotherapeutic agents and strategies for most children with these devastating tumors.
The treatment of low-grade astrocytomas, which cannot be completely excised, has likewise shown only disappointing progress. The chemotherapy agents currently in use have high failure rates and some children suffer multiple, neurologically damaging, relapses. Radiotherapy with its improved conformational techniques is perhaps being delayed too long in some children.
 Prognosis
Prognosis
Despite improvements in surgery and radiation therapy, the outlook for cure remains poor for children with high-grade glial tumors. For children with high-grade gliomas, an early CCG study showed a 45% progression-free survival rate for children who received radiation therapy and chemotherapy, but this may have been due to the inclusion of low-grade patients. More recent studies would suggest survival rate of less than 10%. The major exception to this is congenital glioblastomas which appear to have a much more favorable prognosis. Biologic factors that may affect survival are being increasingly recognized. The prognosis for diffuse pontine gliomas remains very poor, with the standard therapy of radiation alone, being only palliative.
The 5- and even 10-year survival rate for low-grade astrocytomas of childhood is 60%–90%. However, prognosis depends on both site, grade and, it is increasingly realized, on biology. A child with a pilocytic astrocytoma of the cerebellum has a considerably better prognosis than a child with a fibrillary astrocytoma of the cerebral cortex. Pilomyxoid astrocytomas have been recently designated a grade II tumor and have a worse prognosis. For recurrent or progressive low-grade astrocytoma of childhood, relatively moderate chemotherapy may improve the likelihood of survival.
Conventional craniospinal irradiation for children with low-stage medulloblastoma results in survival rates of 60%–90%. Ten-year survival rates are lower (40%–60%). Chemotherapy allows a reduction in the craniospinal radiation dose while improving survival rates for average-risk patients (86% survival at 5 years on the most recent COG average-risk protocol). However, even reduced-dose craniospinal irradiation has an adverse effect on intellect, especially in children younger than age 7 years. Five-year survival rates for high-risk medulloblastoma have been 25%–40%, but this may be improved with the introduction of more chemotherapy during radiation although this still awaits the results of formal trials.
The previously poor prognosis for children with AT/RTs seems improved by intensive multimodality therapy. A single center study suggests an improved outcome for ependymoma using conformational radiation techniques. This awaits confirmation by a national study.
Major challenges remain in treating brain tumors in children younger than age 3 years and in treating brainstem gliomas and malignant gliomas. The increasing emphasis is on the quality of life of survivors, not just the survival rate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


