Source of bilirubin. Bilirubin is derived from the breakdown of heme-containing proteins in the reticuloendothelial system. The normal newborn produces 6 to 10 mg of bilirubin/kg/day, as opposed to the production of 3 to 4 mg/kg/day in the adult.
The major heme-containing protein is red blood cell (RBC) hemoglobin. Hemoglobin released from senescent RBCs in the reticuloendothelial system is the source of 75% of all bilirubin production. One gram of hemoglobin produces 34 mg of bilirubin. Accelerated release of hemoglobin from RBCs is the cause of hyperbilirubinemia in isoimmunization (e.g., Rh and ABO incompatibility), erythrocyte biochemical abnormalities (e.g., glucose-6-phosphate dehydrogenase [G6PD] and pyruvate kinase deficiencies), abnormal erythrocyte morphology (e.g., hereditary spherocytosis [HS]), sequestered blood (e.g., bruising and cephalohematoma), and polycythemia.
The other 25% of bilirubin is called early-labeled bilirubin. It is derived from hemoglobin released by ineffective erythropoiesis in the bone marrow, from other heme-containing proteins in tissues (e.g., myoglobin, cytochromes, catalase, and peroxidase), and from free heme.
Bilirubin metabolism. The heme ring from heme-containing proteins is oxidized in reticuloendothelial cells to biliverdin by the microsomal enzyme heme oxygenase. This reaction releases carbon monoxide (CO) (excreted from the lung) and iron (reutilized). Biliverdin is then reduced to bilirubin by the enzyme biliverdin reductase. Catabolism of 1 mol of hemoglobin produces 1 mol each of CO and bilirubin. Increased bilirubin production, as measured by CO excretion rates, accounts for the higher bilirubin levels seen in Asian, Native American, and Greek infants.
Transport. Bilirubin is nonpolar, insoluble in water, and is transported to liver cells bound to serum albumin. Bilirubin bound to albumin does not usually enter the central nervous system (CNS) and is thought to be nontoxic. Displacement of bilirubin from albumin by drugs, such as the sulfonamides, or by free fatty acids (FFAs) at high molar ratios of FFA:albumin, may increase bilirubin toxicity (see Table 26.1).
Table 26.1 Drugs That Cause Significant Displacement of Bilirubin from Albumin In vitro
Sulfonamides
Moxalactam
Ticarcillin, azlocillin, carbenicillin
Ceftriaxone; cefotetan; cefmetazole, cefonicid
Fusidic acid
Radiographic contrast media for cholangiography (sodium iodipamide, sodium ipodate, iopanoic acid, meglumine loglycamate)
Benzyl alcohol (preservative), benzoate
Aspirin
Ibuprofen
Aminophylline
Diatrizoate
Apazone
Tolbutamide
Rapid infusions of albumin preservatives (sodium caprylate and N-acetyltryptophan)
Rapid infusions of ampicillin
Long-chain FFAs at high molar ratios of FFA:albumin
FFA = free fatty acid. Sources: From Roth P, Polin RA. Controversial topics in kernicterus. Clin Perinatol 1988;15:965-990 and Robertson A, Karp W, Broderson R. Bilirubin displacing effect of drugs used in neonatology. Acta Paediatr Scand 1991;80:1119-1127.
Uptake. Nonpolar, fat-soluble bilirubin (dissociated from albumin) crosses the hepatocyte plasma membrane and is bound mainly to cytoplasmic ligandin (Y protein) for transport to the smooth endoplasmic reticulum. Phenobarbital increases the concentration of ligandin.
Conjugation. Unconjugated (indirect) bilirubin (UCB) is converted to watersoluble conjugated (direct) bilirubin (CB) in the smooth endoplasmic reticulum by uridine diphosphogluconurate glucuronosyltransferase (UGT).
This enzyme is inducible by phenobarbital and catalyzes the formation of bilirubin monoglucuronide. The monoglucuronide may be further conjugated to bilirubin diglucuronide. Both monoglucuronide and diglucuronide forms of CB are able to be excreted into the bile canaliculi against a concentration gradient.
Inherited deficiencies and polymorphisms of the conjugating enzyme gene can cause severe hyperbilirubinemia in neonates. Bilirubin uridine diphosphogluconurate glucuronosyltransferase gene (UGT1A1) polymorphisms have been described, which diminish the expression of the UGT enzyme. The TATA box mutation is the most common mutation found and is implicated in Gilbert syndrome in the Western population. Instead of the usual six (TA) repeats in the promoter region, there is an extra two-base pair (TA) repeat resulting in seven (TA) repeats ([TA]7TAA). The estimated allele frequency among whites is 0.33 to 0.4, and among Asians, it is 0.15. Alone, this mutation may not result in significant neonatal hyperbilirubinemia; however, with other risk factors for hyperbilirubinemia present (G6PD deficiency, ABO incompatibility, HS, and breast milk jaundice), the presence of this mutation may confer a significant risk for neonatal hyperbilirubinemia. The 211G → A (G71R) mutation occurs with increased frequency among the Japanese population, and the presence of this mutation alone (homozygote or heterozygote) can result in reduced enzyme activity and neonatal hyperbilirubinemia. This mutation is also the most common mutation in Japanese patients with Gilbert syndrome. The G71R mutation has not been found in the white population. Other mutations have been described, such as 1456T → G and the CAT box mutation (CCAAT → GTGCT); however, less is known about these mutations and their role in hyperbilirubinemia in the newborn. The population differences in allele frequencies likely account for some of the racial and ethnic variation seen in the development of jaundice.
Excretion. CB in the biliary tree enters the gastrointestinal (GI) tract and is then eliminated in the stool, which contains large amounts of bilirubin. CB is not normally resorbed from the bowel unless it is converted back to UCB by the intestinal enzyme β-glucuronidase. Resorption of bilirubin from the GI tract and delivery back to the liver for reconjugation is called enterohepatic circulation. Intestinal bacteria can prevent enterohepatic circulation of bilirubin by converting CB to urobilinoids, which are not substrates for β-glucuronidase. Pathologic conditions leading to increased enterohepatic circulation include decreased enteral intake, intestinal atresias, meconium ileus, and Hirschsprung disease.
Fetal bilirubin metabolism. Most UCB formed by the fetus is cleared by the placenta into the maternal circulation. Formation of CB is limited in the fetus because of decreased fetal hepatic blood flow, decreased hepatic ligandin, and decreased UGT activity. The small amount of CB excreted into the fetal gut is usually hydrolyzed by β-glucuronidase and resorbed. Bilirubin is normally found in amniotic fluid by 12 weeks’ gestation and is usually gone by 37 weeks’ gestation. Increased amniotic fluid bilirubin is found in hemolytic disease of the newborn and in fetal intestinal obstruction below the bile ducts.
full-term infants to a peak of 6 to 8 mg/dL by 3 to 5 days of age and then falls. A rise to 12 mg/dL is in the physiologic range. In premature infants, the peak may be 10 to 12 mg/dL on the fifth day of life, possibly rising > 15 mg/dL without any specific abnormality of bilirubin metabolism. Levels <2 mg/dL may not be seen until 1 month of age in both full term and premature infants. This “normal jaundice” is attributed to the following mechanisms:
Increased bilirubin production due to:
Increased RBC volume per kilogram and decreased RBC survival (90 days versus 120 days) in infants compared with adults.
Increased ineffective erythropoiesis and increased turnover of nonhemoglobin heme proteins.
Increased enterohepatic circulation caused by high levels of intestinal β-glucuronidase, preponderance of bilirubin monoglucuronide rather than diglucuronide, decreased intestinal bacteria, and decreased gut motility with poor evacuation of bilirubin-laden meconium.
Defective uptake of bilirubin from plasma caused by decreased ligandin and binding of ligandin by other anions.
Defective conjugation due to decreased UGT activity.
Decreased hepatic excretion of bilirubin.
Onset of jaundice before 24 hours of age.
Any elevation of serum bilirubin that requires phototherapy (see Figs. 26.2 and 26.4 and VI.D.).
A rise in serum bilirubin levels of >0.2 mg/dL/hour.
Signs of underlying illness in any infant (vomiting, lethargy, poor feeding, excessive weight loss, apnea, tachypnea, or temperature instability).
Jaundice persisting after 8 days in a term infant or after 14 days in a premature infant.
History
A family history of jaundice, anemia, splenectomy, or early gallbladder disease suggests hereditary hemolytic anemia (e.g., spherocytosis, G6PD deficiency).
A family history of liver disease may suggest galactosemia, α1-antitrypsin deficiency, tyrosinosis, hypermethioninemia, Gilbert disease, Crigler-Najjar syndrome types I and II, or cystic fibrosis.
Ethnic or geographic origin associated with hyperbilirubinemia (East Asian, Greek, and American Indian) (see I.B.3. for potential genetic influences).
A sibling with jaundice or anemia may suggest blood group incompatibility, breast milk jaundice, or Lucey-Driscoll syndrome.
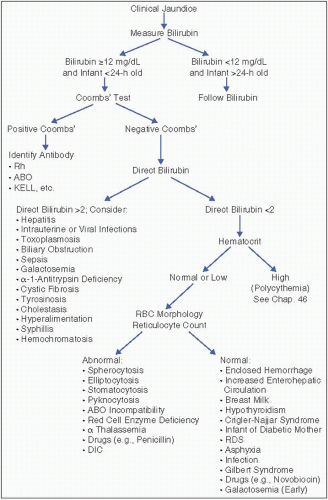
Figure 26.1. Diagnosis of the etiology of hyperbilirubinemia. Rh = rhesus factor; RBCs = red blood cells; DIC = disseminated intravascular coagulation; RDS = respiratory distress syndrome.
Table 26.2 Causes of Neonatal Hyperbilirubinemia
Overproduction
Undersecretion
Mixed
Uncertain mechanism
Fetomaternal blood group incompatibility (e.g., Rh, ABO)
Hereditary spherocytosis (MCHC >36.0 g/dL), eliptocytosis, somatocytosis
Nonspherocytic hemolytic anemias
G6PD deficiency and drugs
Pyruvate-kinase deficiency
Other red cell enzyme deficiencies
α Thalassemia
δ-β Thalassemia
Acquired hemolysis due to vitamin K, nitrofurantonin, sulfonamides, antimalarials, penicillin, oxytocin, bupivacaine, or infection
Metabolic and endocrine conditions
Sepsis
Intrauterine infections
Toxoplasmosis
Rubella
CID
Herpes simplex
Syphilis
Hepatitis
Respiratory distress syndrome
Hypoxia-ischemia
Infant of diabetic mother
Severe erythroblastosis fetalis
Chinese, Japanese, Korean, and American Indian infants (see polymorphism discussion, section I.B.3.)
Breast milk jaundice
Galactosemia
Familial nonhemolytic jaundice types 1 and 2 (Crigler-Najjar syndrome)
Gilbert disease
Hypothyroidism
Tyrosinosis
Hypermethioninemia
Drugs and hormones
Novobiocin
Pregnanediol
Lucey-Driscoll syndrome
Infants of diabetic mothers
Prematurity
Hypopituitarism and anencephaly
Extravascular blood
Petechiae
Hematomas
Pulmonary, cerebral, or occult hemorrhage
Obstructive disorders
Biliary atresia*
Dubin-Johnson and Rotor syndrome*
Choledochal cyst*
Cystic fibrosis (inspissated bile)*
Tumor* or band* (extrinsic obstruction)
α1-antitrypsin deficiency*
Polycythemia
Fetomaternal or fetofetal transfusion
Delayed clamping of the umbilical cord
Increased enterohepatic circulation
Pyloric stenosis*
Intestinal atresia or stenosis, including annular pancreas
Hirschsprung disease
Meconium ileus and/or meconium plug syndrome
Fasting or hypoperistalsis from other causes
Drug-induced paralytic ileus (hexamethonium)
Swallowed blood
Parenteral nutrition
MCHC = mean corpuscular hemoglobin concentration; G6PD = glucose-6-phosphate dehydrogenase; CID = cytomegalovirus inclusion disease, as in TORCH (toxoplasmosis, other, rubella, cytomegalovirus, herpes simplex).
* Jaundice may not be seen in the neonatal period.
Source: Modified from Odell GB, Poland RL, Nostrea E Jr. Neonatal hyperbilirubinemia. In: Klaus MH, Fanaroff A, eds. Care of the High-Risk Neonate. Philadelphia: WB Saunders, 1973, Chapter 11.
Table 26.3 Timing of Follow-up
Infant discharged
Should be seen by age
Before age 24 h
72 h
Between 24 and 47.9 h
96 h
Between 48 and 72 h
120 h
For some newborns discharged before 48 h, two follow-up visits may be required, the first visit between 24 and 72 h and the second between 72 and 120 h. Clinical judgment should be used in determining follow-up. Earlier or more frequent follow-up should be provided for those who have risk factors for hyperbilirubinemia (Table 26.4), whereas those discharged with few or no risk factors can be seen after longer intervals. Source: Reprinted with permission from the American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004;114:297-316.
Maternal illness during pregnancy may suggest congenital viral infection or toxoplasmosis. Infants of diabetic mothers tend to develop hyperbilirubinemia (see Chap. 2).
Maternal drugs may interfere with bilirubin binding to albumin, making bilirubin toxic at relatively low levels (sulfonamides) or may cause hemolysis in a G6PD-deficient infant (sulfonamides, nitrofurantoin, antimalarials).
The labor and delivery history may show trauma associated with extravascular bleeding and hemolysis. Oxytocin use may be associated with neonatal hyperbilirubinemia, although this is controversial. Infants with hypoxic-ischemic injuries may have elevated bilirubin levels; causes include inability of the liver to process bilirubin and intracranial hemorrhage. Delayed cord clamping may be associated with neonatal polycythemia and increased bilirubin load.
Table 26.4 Important Risk Factors for Severe Hyperbilirubinemia
Predischarge TSB or TcB measurement in high-risk or high-intermediate zone
Lower gestational age
Exclusive breastfeeding, especially if it is not going well and infant has excessive weight loss
Jaundice in the first 24 hours of age
Isoimmune or other hemolytic disease
Previous sibling with jaundice
Cephalohematoma or significant bruising
East Asian race
Source: Maisels MJ, Bhutani VK, Bogen D, et al. Hyperbilirubinemia in the newborn infant ≥35 weeks’ gestation: an update with clarifications. Pediatrics 2009;124:1193-1198.
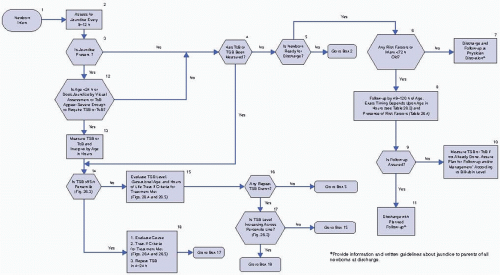
Figure 26.2. Algorithm providing recommendations for management and follow-up according to predischarge bilirubin measurements, gestation, and risk factors for subsequent hyperbilirubinemia. (Reprinted with permission from Maisels MJ, Bhutani VK, Bogen D, et al. Hyperbilirubinemia in the newborn infant ≥35 weeks’ gestation: an update with clarifications. Pediatrics 2009;123:1193—1198.)
Get Clinical Tree app for offline access


