Narcolepsy
Introduction
In 1880, Gelineau coined the term narcolepsie to describe a pathologic condition that was characterized by recurrent, brief attacks of sleepiness.1 He recognized that the disorder was accompanied by falls or astasias, that were subsequently termed cataplexy. Narcolepsy is a lifelong neurologic disorder of rapid eye movement (REM) sleep in which there are attacks of irresistible daytime sleepiness, cataplexy (sudden loss of muscle control in the legs, trunk, face or neck in response to emotional stimuli such as laughter, fright, anticipation of reward or rage), hypnagogic hallucinations (vivid dreams at sleep onset), sleep paralysis (momentary inability to move at the time of sleep onset), and fragmented night sleep.2 There are two main forms of the disorder: narcolepsy with cataplexy and narcolepsy without cataplexy. This chapter provides an overview of childhood narcolepsy.
Epidemiology
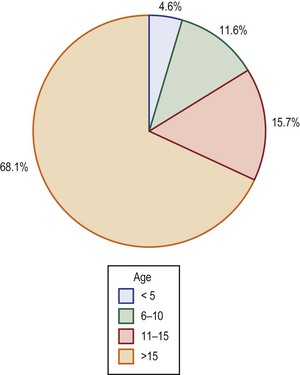
In a community-based survey in Olmsted County, Minnesota, the incidence of narcolepsy was 1.37 per 100 000 persons per year – 1.72 for men and 1.05 for women.3 It was highest in the second decade of life, followed by a gradual decline. The prevalence was approximately 56 persons per 100 000 persons. In Japan, the prevalence has been estimated at 1 in 600,4 and in Israel at 1 in 500 000.5 In Norway, the prevalence of narcolepsy with cataplexy has been estimated at 0.022% based on a survey of 20- to 60-year-olds.6 The exact prevalence rate of narcolepsy in childhood has been difficult to establish. Some epidemiologic studies have required the presence of cataplexy as a prerequisite for the diagnosis,7 whereas others8 have not made this stipulation. This lack of uniformity in clinical diagnostic criteria may explain variability in estimations of the prevalence of narcolepsy.9 There seems to be a slight male predominance for prevalence – in the Olmsted County study, the male:female ratio was 1.8 : 1.3 Narcolepsy has been recognized as early as 1 year of age, though most subjects tend to be adolescents at the time of diagnosis. A seasonal variation in the incidence of narcolepsy has been observed in China, with the lowest incidence in November, and the highest incidence in April.10 Further, the data from China also indicate an almost threefold increase in the incidence of narcolepsy following the 2009 H1N1 influenza pandemic, which might suggest a role for immune-mediated dysfunction consequent to this infection in the pathogenesis of the disorder.10 Although the disorder is most often diagnosed in the third and fourth decades, a meta-analysis of 235 subjects derived from three studies by Challamel and coworkers10 found that 34% of all subjects had onset of symptoms prior to the age of 15 years, 16% prior to age 10 years, and 4.5% prior to age 5 years (Figure 18-1). Increasing awareness of narcolepsy in childhood and the avaiability of cerebrospinal fluid hypocretin testing is likely leading to diagnosis at progressively earlier ages, including in preschool-aged children. A lag period of 5 to 10 years between the onset of symptoms and diagnosis has however been observed in adult subjects.11 Cataplexy, the most specific clinical feature of narcolepsy, is present in only 50% to 70% of all subjects.
Clinical Features
Preschool-Aged Children
In their meta-analysis of 235 children, Challamel and colleagues found that 4.6% were below the age of 5 years at the time of diagnosis.10 Sharp and D’Cruz have described a 12-month-old with hypersomnia who was subsequently confirmed to have narcolepsy.12 In general, it is difficult to diagnose narcolepsy prior to the age of 4 to 5 years, as even unaffected children of this age tend to take habitual daytime naps and are not able to provide an accurate history of cataplexy, hypnagogic hallucinations, or sleep paralysis. The diagnosis may, however, be facilitated by documentation of cataplexy attacks on video-polysomnography, which shows skeletal muscle atonia and bursts of rapid eye movements coinciding with low-voltage, mixed-frequency activity on the electroencephalogram (EEG). The availability of cerebrospinal fluid (CSF) hypocretin analysis also facilitates the diagnosis of narcolepsy–cataplexy in this age group, but this does not apply to narcolepsy without cataplexy, as the latter group is not associated with reduction in CSF hypocretin levels.
School-Aged Children
Daytime sleepiness is an invariant and most disabling feature of narcolepsy. It can manifest itself as early as 5 to 6 years of age. There is a background of a constant, foggy feeling from drowsiness, superimposed on which are periods of more dramatic sleep attacks. Habitual afternoon napping is uncommon in healthy children above age 5 or 6 years, and should raise suspicion of narcolepsy. Lenn reported a 6-year-old who would fall asleep 5 to 10 times a day.13 Wittig and colleagues have described a 7-year, 5-month-old boy with narcolepsy who tended to fall asleep while watching television for longer than a half hour, at the dinner table, and while seated in his mother’s lap at a doctor’s office.14 The naps in children with narcolepsy tend to be longer than those in adult patients (30 to 90 minutes), but they are not consistently followed by a refreshed feeling.15 These attacks of sleepiness are most likely to occur when the patient is carrying out sedentary activities such as sitting in a classroom or reading a book. Sleepiness occurs regardless of the quantity of night sleep. The daytime sleepiness is frequently associated with automatic behavior of which the subject is unaware, impaired consolidation of memory, decreased concentration, executive dysfunction, and school-related learning problems. The irritability and mood swings that accompany sleepiness may mimic depression.16,17 Children with daytime sleepiness may be mistakenly labeled ‘lazy’ and frequently become the target of negative comments from their peers. Excessive sleepiness might also be overlooked by the parents until it starts adversely impacting mood, behavior, or academic performance. These behavioral changes likely reflect impairment of function of the ventro-lateral prefrontal cortex from sleepiness.18 Pollack studied the circadian sleep–wake rhythms in subjects with narcolepsy who were isolated from their environmental cues.19 He found that the major sleep episode was still about 6 hours long, and it occurred about once every 24 hours, thus indicating that the circadian clock was functioning normally. Pollack confirmed that patients with narcolepsy tended to sleep more often, but not longer than people without narcolepsy.
Cataplexy, the second most common but most specific feature of narcolepsy, consists of a sudden loss of muscle tone in the facial muscles, or those of the thighs, back, or neck in response to emotional triggers such as fright, rage, excitement, surprise, or laughter or the anticipation of reward. It is caused by the intrusion of the skeletal muscle atonia of REM sleep into wakefulness.20,21 Cataplexy is associated with hyperpolarization of spinal alpha motor neurons, with resultant active inhibition of skeletal muscle tone and suppression of the monosynaptic H-reflex and tendon reflexes. A history of cataplexy may be difficult to elicit in young children. The author recalls a 6-year-old girl with proven narcolepsy who denied any episodic muscle weakness but would repeatedly fall down whenever she jumped on a trampoline. Consciousness remains fully intact during the cataplexy episodes, which can last 1 to 30 minutes. Respiration and cardiovascular function remain unaffected. Challamel and coworkers10 found cataplexy in 80.5% of idiopathic narcolepsy and in 95% of symptomatic narcolepsy subjects.
Night-time sleep is also disturbed in narcolepsy with frequent awakenings. Young and colleagues attributed sleep fragmentation in part to periodic limb movements, which were found in five (63%) of eight children with narcolepsy in their series.22 Periodic limb movements are rhythmic limb muscle contractions of 0.5 to 5 seconds’ duration, with an intermovement interval of 5 to 120 seconds, occurring in series of three or more, usually during stage 1 or 2 of non-REM (NREM) sleep. They may or may not be associated with EEG evidence of cortical arousal. They may be a marker for underlying restless legs syndrome. Sleep fragmentation in narcolepsy may also occur unrelated to periodic limb movements or restless leg syndrome. Decreased pressure for NREM sleep homeostasis and altered NREM–REM sleep interaction has also been proposed as underlying the nocturnal sleep disruption of narcolepsy.89
Psychosocial problems in children with narcolepsy have been studied.90 They found that, compared to hypersomnolent controls, children with narcolepsy showed more behavioral difficulties, depressed mood and impaired quality of life.91
Obesity may develop at the onset of narcolepsy–cataplexy symptoms, in association with hyperphagia and binge eating. It may be related to loss of the physiological rise in leptin levels at night (leptin acts as an appetite suppressant), or to sedentary behavior and decreased basal metabolism. Obesity does not seem to be related to the use of anticholinergic medications, as it develops even prior to initiation of drug treatment. The obesity may be accompanied by obstructive sleep apnea in 9–19% of subjects.92 Precocious puberty may also be seen in preteen age boys and girls at onset of narcolepsy–cataplexy.
REM sleep behavior can sometimes be a presenting manifestation of narcolepsy.93,94 It is characterized by motor dream enactment, typically in the form of flailing of arms and legs and yelling behavior. Simultaneously obtained polysomnogram shows REM sleep without atonia, i.e., persistence of muscle tone during sleep.
Pathophysiology
The study of narcolepsy in humans has been advanced by the study of the disorder in animals. Narcolepsy has been studied in cats, miniature horses, quarter horses, Brahman bulls, and about 15 breeds of dogs. In animals, it shows a monogenic, autosomal recessive pattern of inheritance.23–25 Cataplexy can be induced in cats by the injection of carbachol (an acetylcholine-like substance) into the pontine reticular formation.26 Specifically, muscarinic type-2 receptors of acetylcholine have been implicated.27,28 The food-elicited cataplexy test, used to study cataplexy in dogs, uses the finding that the time taken for the consumption of food by narcoleptic animals whose eating is interrupted by cataplexy attacks is much longer than in animals without narcolepsy.
In 1999, Lin and coworkers demonstrated that canine narcolepsy is caused by a mutation in the hypocretin receptor-2 (orexin-2) gene.29 Around the same time, Chemelli and colleagues established that a null mutation for the hypocretin-1 and hypocretin-2 peptides in mice produces aspects reminiscent of human narcolepsy, including cataplexy.30 The hypocretin-containing neurons are located primarily in the dorsolateral hypothalamus. They have widespread projections to the basal forebrain, amygdala, medial nuclei of the thalamus, periaqueductal gray matter, reticular formation, pedunculo-pontine nucleus, locus coeruleus, raphe nucleus, pontine tegmentum, and dorsal spinal cord.29,31 Hypocretins 1 and 2 are peptides that are synthesized from preprohypocretin, and have corresponding receptors. Although the hypocretin type 1 receptor binds only to hypocretin-1, the hypocretin type 2 receptor can bind to both type 1 and type 2 ligands. Hypocretins stimulate food intake, increase the basal metabolic rate, and promote arousal.31 Decreased activation of the hypocretin system is the underlying theme in canine and murine narcolepsy. This receptor down-regulation may occur as a result of either exon-skipping mutations in the hypocretin receptors (Labrador and Doberman models),30 or after point mutations in the Hcrt2 receptor gene, with an amino acid change from glutamic acid to lysine in the N-terminus portion of the receptor (Dachshund model).32 Also of significance is the finding that the intravenous administration of hypocretin-1 (orexin A) in narcoleptic Doberman pinschers reduces cataplexy for up to 3 days, increases activity, promotes waking, and reduces sleep fragmentation in a dose-dependent manner.33
Histocompatibility Antigens and Human Narcolepsy
In 1984, an association between narcolepsy and histocompatibility leukocyte antigen (HLA) DR2 was reported in Japan by Juji and coworkers.34 This association was subsequently observed in other geographic regions of the world as well.35,36 Consequently, an immunologic mechanism was suspected in the pathogenesis of human narcolepsy, but this has not been established. It was then demonstrated that the association with DR2 is only secondary, and that there is a stronger association of narcolepsy with the HLA DQ antigens, specifically DQB1*0602 and DQA1*0102, which are present in 95% to 100% of patients, as compared to a 12% to 38% prevalence in the general population.37 In a study of 525 healthy subjects, Mignot and colleagues demonstrated that DQB1*0602 positivity was linked to shorter REM latency, increased sleep efficiency, and decreased time spent in stage 1 NREM sleep.38 Pelin and coworkers have demonstrated that homozygosity for these two haplotypes is associated with a twofold to fourfold increase in the likelihood of developing narcolepsy over heterozygotes, but that the presence of these antigens does not influence the severity of the disease.39
Hypocretins and Human Narcolepsy
Unlike narcolepsy in dogs and mice, human narcolepsy–cataplexy is generally not associated with abnormalities in hypocretin receptors but rather with low to absent levels of cerebrospinal (CSF) hypocretin-1.40 In a postmortem study of human narcolepsy, Thannickal and colleagues found an 85% to 95% reduction in the number of hypocretin neurons in the hypothalamic region,41 whereas melanin-concentrating hormone neurons, which are intermingled with the hypocretin neurons, remained unaffected, thus suggesting a targeted neurodegenerative process. Using a radioimmunoassay, Nishino and coworkers42 found that the mean CSF level of hypocretin-1 in healthy controls was 280.3 ± 33.0 pg/mL, and in neurologic controls it was 260.5 ± 37.1 pg/mL, whereas in those with narcolepsy, hypocretin-1 was either undetectable or below 100 pg/mL. The diagnostic sensitivity of low levels (less than 100 pg/mL) was 84.2%. Low to absent levels were found in 32 out of 38 patients, who were all HLA DQB1*0602 positive. HLA-negative narcolepsy patients had normal to high CSF hypocretin-1 levels. In another recent study, 92.3% of patients who were both DQB1*0602 and cataplexy positive had undetectable CSF hypocretin-1 levels, whereas DQB1*0602-negative patients with cataplexy and DQB1*0602-negative patients without cataplexy had normal levels.43 In a study of narcolepsy with cataplexy, narcolepsy without cataplexy and idiopathic hypersomnia, Kanbayashi et al.44 found that all nine patients who were hypocretin deficient in the cerebrospinal fluid (CSF) were HLA DR2 (i.e. HLA DQB1*0602) positive. In contrast, narcolepsy without cataplexy and idiopathic hypersomnia were associated with normal levels of CSF hypocretin (Figure 18-2).
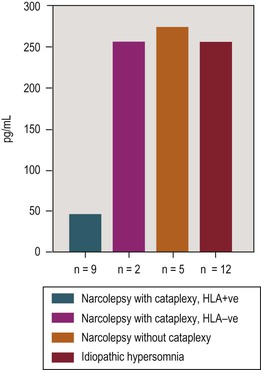
Figure 18-2 Mean cerebrospinal fluid hypocretin-1 levels in various categories of hypersomnia. The narcolepsy-cataplexy group included three prepubertal children of ages 6, 7, and 10. Adapted from Kanbayashi T, Inoue Y, Chiba S, et al: CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. J Sleep Res 2002;11:91–93.
The Two-Hit Hypothesis
The presence of histocompatibility antigen DQB1*0602 is, per se, insufficient to precipitate narcolepsy. This is substantiated by the fact that DQB1*0602-positive monozygotic twins have been incompletely concordant for narcolepsy, with one of the pair developing narcolepsy–cataplexy at age 12 years and the other not until after having suffered emotional stress and sleep deprivation at the age of 45 years.45 Human narcolepsy is therefore best explained on the basis of a two-threshold hypothesis, with an interplay between genetic susceptibility and environmental factors – major life events, such as a systemic illness, an injury, or bereavement, have been reported to be present in 82% of narcolepsy patients, compared with a 44% incidence in controls46 (P <0.001). The combination of genetic susceptibility and an acquired stress seems to trigger most cases of narcolepsy. Recently, there has been documentation of narcolepsy–cataplexy development following H1N1 influenza infection or immunization in children and adults who were positive for the HLA DQB1*0602 antigen.95
Monoamine Disturbances
Hypocretin deficiency in humans leads to down-regulation of arousal-mediating noradrenergic and dopaminergic pathways in the brainstem, and to up-regulation of REM sleep-facilitating cholinergic pathways.47 Montplaisir and coworkers48 measured serum and CSF levels of several biogenic amines and their metabolites – dopamine (the metabolite homovanillic acid), norepinephrine (the metabolite 3-methoxy-4-hydroxyphenylethyleneglycol), epinephrine, and serotonin (the metabolite 5-hydroxy indoleacetic acid) – in patients with narcolepsy, in those with idiopathic hypersomnia, and in normal controls. Both narcolepsy and idiopathic hypersomnia patients had significantly decreased concentrations of dopamine and indoleacetic acid, a metabolite of tryptamine. Dopamine and tryptamine are usually present in high concentrations in the basal ganglia. A relative deficiency of these compounds, probably mediated by down-regulation of hypocretin, is involved in the evolution of sleepiness. Stimulants such as dextroamphetamine and methylphenidate, which are used in the treatment of hypersomnolence, are known to enhance dopamine release from presynaptic terminals. Activation of selective dopamine D2 receptor agonists also suppresses cataplexy.49 This inhibition is believed to be indirectly mediated via activation of noradrenergic pathways. A reduction in central dopamine activity might also underlie the periodic leg movements that are common in the night sleep of patients with narcolepsy; they are usually relieved by treatment with levodopa or dopamine agonists. A cholinergic disturbance also coexists; physostigmine, a cholinergic agent, leads to a transient increase in cataplexy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


