Myopathies
Basil T. Darras
MUSCULAR DYSTROPHIES
Muscular dystrophies are an inherited group of primary diseases of muscle, characterized pathologically by muscle fiber degeneration and clinically by progressive muscle weakness. Pathologic, clinical and genetic criteria have been used as the basis for their classification.
DUCHENNE/BECKER MUSCULAR DYSTROPHIES (DYSTROPHINOPATHIES)
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are progressive myopathies, inherited as X-linked recessive traits. DMD is the most common form of muscular dystrophy, with an incidence of about 1 in 3300 live male births and a prevalence rate in the total population of about 3 per 100,000. BMD has a similar presentation but a relatively milder clinical course. The reported incidence of BMD has varied from about 1 in 18,000 to 1 in 31,000 male births. In addition, there is an intermediate group of patients with either mild DMD or severe BMD, who are known as outliers. It is now well known that all 3 types of muscular dystrophy are allelic, resulting from dystrophin deficiency due to mutations of a single gene, called the dystrophin gene.1
Other dystrophinopathies, occurring at a lower incidence, include:
• manifesting DMD/BMD carrier females,
• X-linked dilated cardiomyopathy, and
• muscle cramps with myoglobinuria.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Great heterogeneity in the clinical features and course of the various dystrophinopathies has been observed, creating a spectrum ranging from very mild to very severe presentations. The severe end of the spectrum includes DMD, BMD the outliers or intermediate phenotype in which skeletal muscle is primarily affected, and X-linked dilated cardiomyopathy in which the heart is the organ primarily affected. Females who carry DMD/BMD can be totally asymptomatic or can manifest mild to severe symptoms.
Clinically, the distinction between DMD and BMD is made by the age of wheelchair confinement, which is less than 13 years in DMD and beyond 16 years in BMD. Patients who become wheelchair-bound between 13 years and 16 years are classified as outliers or as exhibiting an intermediate presentation. The outlier group could be classified clinically as having either mild DMD or severe BMD.
Duchenne Muscular Dystrophy
In children with Duchenne muscular dystrophy (DMD), although there is histologic and laboratory evidence of myopathy from birth, the onset of weakness usually occurs between 2 and 3 years of age; it may be delayed and become apparent after the age of 3 years, but almost all patients with DMD become symptomatic before age 5 years. The child usually has difficulty with running, jumping, going up steps, and other similar activities; an unusual waddling gait, lumbar lordosis, and calf enlargement are usually observed. Muscular weakness is symmetrical and selectively affects proximal limb muscles before distal and the lower extremities before the upper. Early on, the patient may complain of leg pains. Jumping and running are almost impossible in most cases, and, in arising from the floor, affected boys use hand support to push themselves to an upright position (Gower sign). Neck flexor weakness occurs at all stages of the disease and distinguishes boys with DMD from patients with milder presentations; at least early on, patients with BMD and outliers appear to have preserved neck flexor strength. Cardiac muscle is also affected. Most children with DMD often have varying degrees of nonprogressive impairment of cognitive function, although an occasional child may have average or above-average intelligence. Earlier reports suggested that verbal IQ was more affected than performance IQ. Recently, however, a more specific cognitive profile has emerged, demonstrating deficits in working memory and executive function.
Physical examination shows pseudohyper-trophy of the calf muscles (Fig. 572-1) and, in some instances, of quadriceps, gluteal, deltoid, and other muscles, and lumbar lordosis, waddling gait, shortening of the Achilles tendons, and hyporeflexia or areflexia. The shortening of the Achilles tendons leads to toe walking.
Between 3 and 6 years of age, there may be some evidence of transient improvement, known as the “honeymoon phase” of muscular dystrophy; this is gradually followed by relentless deterioration, leading to wheelchair confinement by the age of approximately 13 years. Wheelchair-bound children tend to develop contractures and scoliosis, with deterioration of pulmonary function. The incidence of cardiomyopathy increases gradually in teenage years, with about one third of patients being affected by age 14 years, one half by age 18 years, and all patients after age 18 years. Intestinal hypomotility, also known as intestinal pseudo-obstruction, is an important and sometimes life-threatening complication in patients with Duchenne muscular dystrophy (DMD). It may present with acute gastric dilatation, vomiting, and abdominal pain and distention and seems to be related to smooth muscle degeneration. Most patients with DMD die in their late teens or twenties (mean: 20 ± 3.9 years) from respiratory insufficiency or from cardiac failure secondary to progressive cardiomyopathy (10–40%). In some, the immediate cause of death is not apparent.2 Assisted ventilation can prolong life expectancy, but the patient will be dependent for activities of daily living.
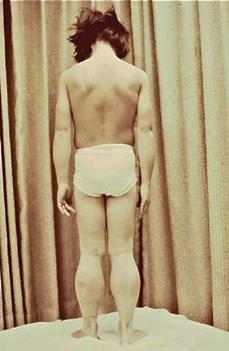
FIGURE 572-1. Pseudohypertrophy of the calf muscles in a patient with Duchenne muscular dystrophy. (Courtesy of Theodore Munsat, MD, New England Medical Center, Boston MA. Reprinted from: Darras BT, Menache CC, Kunkel LM. Dystrophinopathies. In: Jones HR Jr, De Vivo DC, Darras BT, eds. Neuromuscular Disorders in Infancy, Childhood, and Adolescence: A Clinician’s Approach. Philadelphia, PA: Butterworth-Heinemann; 2003:649; with permission from Elsevier.)
Becker Muscular Dystrophy
In Becker muscular dystrophy, the age of onset of symptoms is usually later, between the ages of 5 and 15 years (mean: approximately 12 years) or sometimes even in the third or fourth decade or later, and the degree of clinical involvement is milder; cardiac disease and cognitive impairment are not as common or as severe as in the Duchenne variety, and gastrointestinal symptoms are essentially absent. Also, contractures and scoliosis are not as likely to develop in BMD. In addition, in Becker and intermediate types of muscular dystrophy, neck flexor muscle strength is relatively well preserved. Patients with BMD typically remain ambulatory beyond the age of 16 years and into adult life; they usually survive beyond the age of 30 years, with death from respiratory failure or cardiomyopathy/cor pulmonale usually occurring between 30 and 60 years.3Mean age at death is in the mid-40s. Serum creatine kinase levels are usually markedly increased in Becker dystrophy and, therefore, cannot be used as a way of differentiating between the 2 types of dystrophy.
Manifesting Duchenne Muscular Dystrophy (DMD)/Becker Muscular Dystrophy (BMD) Carrier Females
Carriers are usually free of symptoms (DMD, 76%; BMD, 81%) but may have mildly increased serum creatine kinase and usually mild calf hypertrophy. However, 8% to 19% present with mild to moderate, but occasionally severe, muscle weakness of the limb-girdle type or even with DMD/BMD.
 GENETICS
GENETICS
Dystrophin Gene
The DMD/BMD gene, now known as the dystrophin gene, is the largest gene yet identified in humans, spanning approximately 2.3 megabases on the short arm of the X chromosome at Xp21.1 The protein product dystrophin has a total molecular weight of 427 kilodaltons (kD) and is recognized on Western blots of human skeletal muscle proteins using antidystrophin antibodies.4 With the use of immunocytochemistry, dystrophin has been localized to the cytoplasmic face of the plasma membrane of muscle fibers. It has also been shown that dystrophin is part of a large, tightly associated glycoprotein complex containing many other proteins (Fig. 572-2). It is believed that in normal cells, the dystrophin stabilizes the glycoprotein complex and protects it from degradation; in the absence of dystrophin, the complex becomes unstable. There is almost always secondary reduction in the amount of proteins of the glycoprotein complex in the muscle tissue of patients with DMD. The loss of associated membrane proteins as a result of dystrophin deficiency may initiate the degenerative changes seen in muscular dystrophy.
Dystrophin Gene Mutations
Of the dystrophin gene mutations identified so far, most are deletions, detected in approximately 65% of DMD patients and 85% of BMD patients.1 Partial gene duplications have also been reported in 6% to 10% percent of patients. In the remaining patients without detectable deletions or duplications, the molecular lesions represent small insertions/deletions, point mutations, or splicing errors (Table 572-1).
Published studies have failed to reveal any apparent correlation between the size of dystrophin gene deletions and the severity and progression of the DMD/BMD phenotype. The molecular basis of Duchenne versus Becker muscular dystrophy seems to be related to the disruption or preservation of the amino acid reading frame by the deletion mutations. The latter either disrupt or preserve the reading frame in most cases of Duchenne or Becker muscular dystrophy, respectively.
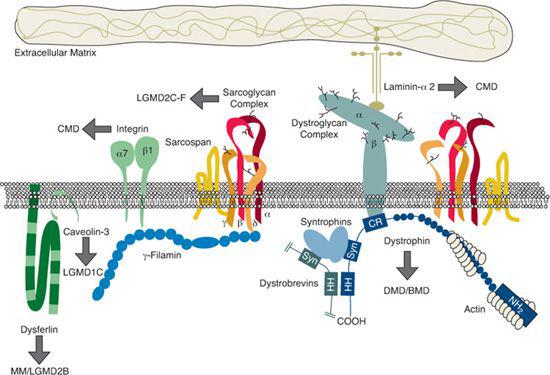
FIGURE 572-2. The dystrophin-associated protein complex. Arrows indicate the protein components mutated in various muscular dystrophies. The laminin a2-chain gene is mutated in a subtype of congenital muscular dystrophy without structural brain anomalies and the sarcoglycan proteins in patients with sarcoglycanopathies (autosomal recessive LGMDs). BMD, Becker muscular dystrophy; CMD, congenital muscular dystrophy; DMD, Duchenne muscular dystrophy; LGMD, limb-girdle muscular dystrophy. (Courtesy K O’Brien and L Kunkel, Children’s Hospital Boston. Reprinted from: Darras BT, Menache CC, Kunkel LM. Dystrophinopathies. In: Jones HR Jr, De Vivo DC, Darras BT, eds. Neuromuscular Disorders in Infancy, Childhood, and Adolescence: A Clinician’s Approach. Philadelphia, PA: Butterworth-Heinemann; 2003:649; with permission from Elsevier.)
Table 572-1. Molecular Genetic Testing Used in the Dystrophinopathies

Dystrophin
More than 99% of DMD patients display complete or almost complete absence of dystrophin in skeletal muscle biopsy specimens. The test is very specific because patients with neuromuscular diseases other than DMD/BMD have normal dystrophin. Patients with dystrophin levels between 5% and 20% of normal, regardless of protein size, seem to develop an intermediate phenotype (mild DMD or severe BMD). Patients with mild to moderate Becker phenotype usually have levels above 20%.4
 DIAGNOSIS
DIAGNOSIS
Before the age of 5 years, the serum creatine kinase levels are usually 10 to 200 times the upper limit of normal, or even higher; levels of 10,000 to 50,000 IU/L are not unusual in Duchenne muscular dystrophy (DMD)/Becker muscular dystrophy (BMD). In a child with DMD, during the first 3 years of life the serum creatine kinase concentration is always more than 10 times the upper limit of normal; if it is less than that, the diagnosis should be questioned.
The muscle biopsy results demonstrate degeneration, regeneration, isolated “opaque” hypertrophic fibers, and significant replacement of muscle by fat and connective tissue. In muscle biopsy tissue derived from DMD patients, there is complete or almost complete absence of staining with antidystrophin antibodies, but in BMD patients either normal or reduced patchy staining of the sarcolemma is observed. In patients with other neuromuscular diseases, there is homogeneous staining of the plasma membrane.1,5
 TREATMENT
TREATMENT
Therapeutic interventions in DMD/BMD are aimed at maintaining function, preventing contractures, and providing psychologic support. Passive stretching exercises to prevent contractures of the iliotibial band, the Achilles tendons, and flexors of the hip are the mainstays of physical therapy. Lightweight plastic ankle-foot orthoses (AFOs) should be applied if the foot remains in plantar flexion during sleep. Standing and/or walking can be maintained by using long leg braces. Surgery can be performed to release contractures of the hip flexors, iliotibial bands, and Achilles tendons. Standing and ambulation seem to prevent scoliosis. After the age of 10 years, pulmonary function studies, electrocardiography, and echocardiography should be performed annually or biannually to monitor the pulmonary and cardiac functions. Overnight mouth intermittent positive pressure can be used to treat symptomatic nocturnal hypoventilation, and respiratory assistance may be used during periods of respiratory infection.
Medications in Duchenne Muscular Dystrophy
Clinical studies have shown that prednisone improves the strength and function of patients with DMD.6-8 Deflazacort is a synthetic derivative of prednisolone used in Europe, but currently unavailable in the United States. It has been suggested that deflazacort has fewer side effects than prednisone, particularly relating to weight gain.9,10
The American Academy of Neurology and the Child Neurology Society have published national practice parameters for the use of corticosteroid therapy; some of the recommendations, which follow, are in accordance with those parameters.11
• Offer boys with DMD, who are older than 5 years, treatment with prednisone at a dose of 0.75 mg/kg/day. Discuss carefully the potential benefits and risks of corticosteroid therapy with the patient and family prior to initiating therapy.
• Assess the potential benefits of corticosteroid therapy.
• Continue the optimal maintenance dose of prednisone, 0.75 mg/kg/day, if side effects are not severe. Gradual tapering of prednisone to as low as 0.3 mg/kg/day supports significant but less robust improvement.
• DMD may also be treated using deflazacort 0.9 mg/kg/day. Monitor side effects of weight gain and asymptomatic cataracts.
Prevention of Secondary Complications
A number of strategies may be employed to prevent secondary complications of DMD/BMD. Patients should be evaluated by pulmonary and cardiac specialists prior to surgery and receive pneumococcal vaccine and annual influenza vaccination.12 They should receive exposure to sunshine as well as a balanced diet rich in vitamin D and calcium to improve bone density and reduce the risk of fractures. If the vitamin D serum concentration is less than 20 ng/mL, vitamin D supplementation may be initiated. Patients should receive physical therapy to promote mobility and prevent contractures and be encouraged to control weight and avoid obesity. Routine evaluation by a nutritionist is recommended.
Surveillance
Individuals with DMD and BMD should receive routine monitoring by a cardiologist for evidence of cardiomyopathy starting at around the age of 10 years, and cardiac evaluation should be repeated annually or biannually.2 Female carriers should be monitored for dilated cardiomyopathy after their teenage years.13 DMD/BMD patients should be monitored for orthopedic complications, especially scoliosis, and receive surgical interventions as needed. Baseline pulmonary function testing should be obtained before wheelchair confinement, at about 9 to 10 years of age. A pediatric pulmonologist should evaluate patients twice a year after any 1 of the following events: wheelchair confinement, vital capacity reduced below 80% predicted, or age 12 years.12
EMERY-DREIFUSS MUSCULAR DYSTROPHY
Emery-Dreifuss muscular dystrophy (EDMD) is an X-linked recessive (chromosome Xq28) or autosomal dominant or autosomal recessive condition (chromosome 1q21) with onset in late childhood or adult life. Mutations in the emerin (Xq28)16 and lamin A/C genes (1q21)17 are responsible for the EDMD form of muscular dystrophy.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The muscle weakness and wasting in EDMD has a humeroperoneal distribution, often starting in the arms, with weakness of both the biceps and triceps and relative preservation of the deltoid muscles.18 Later, distal leg weakness with atrophy of the peroneal muscles is noted; mild facial weakness may be also be observed. The myopathy tends to be slowly progressive. Contractures at the elbows are noted early, often associated with toe-walking, as the first manifestations of the disease. Contractures of the posterior aspect of the neck, lower spine, and the Achilles tendons also occur. Cardiac involvement is common and consists of cardiomyopathy, with atrioventricular block and often atrial paralysis.19 The cardiomyopathy may lead to sudden death in approximately 50% of the affected individuals, usually early in adult life.
 DIAGNOSIS
DIAGNOSIS
Laboratory studies show modest elevation of serum creatine kinase levels, which are rarely above a few hundred units per liter. DNA testing for emerin and lamin A/C gene mutations is now available. A muscle biopsy can provide further evidence for the diagnosis of X-linked EDMD by demonstrating absence of nuclear immuno-staining for emerin by immunohistochemistry.
 TREATMENT
TREATMENT
Because the cardiac involvement in Emery-Dreifuss muscular dystrophy is potentially fatal, the cardiac status of the patient should be investigated even if he/she is asymptomatic. Installation of a cardiac pacemaker may be lifesaving in patients with evidence of atrioventricular block. Stretching exercises to prevent the development of contractures should be the focus of physical therapy.
MYOTONIC DYSTROPHY TYPE 1
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy among those of European ancestry, with a prevalence of 3 to 5 per 100,000 population and an incidence of 1 in 8000. DM1 is a multisystem disorder, transmitted by autosomal dominant inheritance, with variable penetrance. In the classical form, DM1 has its onset in adolescence or adulthood, but a neonatal form also occurs. The main clinical features of DM1 are myotonia (delayed muscle relaxation after contraction), weakness and wasting affecting facial and distal limb muscles, frontal balding (in males), cataracts, cardiomyopathy with conduction defects, multiple endocrinopathies, hypersomnia, and low intelligence or dementia. The face is long, with wasting of the masseter and temporal muscles and variable ptosis and facial diplegia. There may be associated dysarthria, hearing loss, swallowing difficulties, and mild external ophthalmoplegia. Myotonia can be an early symptom, demonstrated by percussion of muscles, usually of the thenar eminence, and by difficulty with releasing the grasp or opening the eyes after sustained closure. Later in the course of the disease, the progressive muscle weakness and wasting become the predominant features, leading to severe distal weakness in the hands and feet. Endocrinopathies include hyperinsulinism, rarely diabetes, adrenal atrophy, hypothyroidism, infertility in women, testicular atrophy, and growth hormone secretion disturbances. Smooth and cardiac muscle involvement are usually expressed by disturbed gastrointestinal mobility and cardiac conduction defects.20
Fifteen percent to 25% of offspring of affected DM1 mothers develop congenital myo-tonic dystrophy. The congenital form of DM1 presents with profound hypotonia at birth, associated with facial diplegia, feeding and respiratory difficulties, and skeletal deformities such as clubfeet. Later, during childhood, delayed developmental progression is noted in a pattern consistent with mental retardation. Clinical myotonia is usually absent in neonates and infants. Brain imaging reveals ventriculomegaly due to brain atrophy.
Although cardiac arrhythmia and atrioventricular block have been thought to be rare in DM1, a retrospective study found that children with the congenital or infantile form of DM1, and asymptomatic adolescents with no or only subtle signs of myotonic dystrophy experienced serious cardiac rhythm disturbances as early as the second decade of life.21 Thus it has been recommended that the routine evaluation of young patients with DM1 include exercise testing with EKG monitoring.
 DIAGNOSIS
DIAGNOSIS
The identification of the myotonic dystrophy mutation has provided a molecular blood test for almost 100% accurate diagnosis of this disorder in both symptomatic and asymptomatic individuals.
The DM1 locus was mapped by linkage analysis to chromosome 19q13.3; this genetic localization finally led to the recent identification of the genetic defect in DM type 1, which is thought to be an amplified trinucleo-tide CTG repeat, located in the 3′ untranslated region of a gene that putatively encodes a serine-threonine protein kinase (myotonin-protein kinase; DMK).22 Although this CTG repeat is quite polymorphic, it is stable in normal individuals. In contrast, the CTG repeat in DM1 chromosomes is unstable and can become significantly large. In normal individuals, the 2 alleles contain between 5 and 50 copies of the CTG repeat. However, normal individuals with 38 to 49 copies of the repeat are classified in a borderline or pre-mutation category because of the small possibility of expansion of the CTG repeat in their offspring or family members. Mildly affected individuals or asymptomatic protomutation “carriers” have 50 to 80 CTG repeats, whereas symptomatic subjects have between 80 and 2000 or more copies (full mutation). 
 TREATMENT
TREATMENT
The treatment of myotonic dystrophy is currently symptomatic. As patients develop distal weakness, braces for foot drop are usually helpful. The myotonia frequently responds to medications that stabilize membranes, such as phenytoin, mexiletine, gabapentin, carbamazepine, and acetazolamide.
Neonates with congenital myotonic dystrophy type 1 (DM1) often require continuous ventilatory support. Support required longer than 4 weeks usually indicates a poor prognosis for survival. During the first 2 years of life, feeding difficulties are common; children with congenital DM1 are at increased risk for aspiration and may benefit from feeding evaluation. During the first 6 months of life, gastrostomy tube insertion is often necessary to maintain nutrition and prevent aspiration pneumonia. Clubfoot deformities require orthopedic care.
MYOTONIC DYSTROPHY TYPE 2
During the last 15 years, a subgroup of families with myotonia, weakness, and cataracts, but with other features atypical for DM, were identified because they had no abnormal expansion of the CTG repeat in the DM gene on chromosome 19. Most patients are adults. Myotonic dystrophy type 2 is also known as proximal myotonic myopathy (PROMM).24
LIMB-GIRDLE MUSCULAR DYSTROPHIES
The term limb-girdle dystrophy embraces a number of conditions with heterogeneous etiologies; a European Neuromuscular Center meeting in 1995 defined limb-girdle muscular dystrophy (LGMD) as a muscular dystrophy with predominantly proximal distribution of weakness that, early in the course of the disease, spares distal muscles as well as facial and extraocular muscles.25 Most cases are inherited in an autosomal recessive fashion and, as is to be expected, are sporadic. However, families with an autosomal dominant pattern of inheritance have also been described. The discovery of the genetically distinct subtypes of LGMD has led to nomenclature designating autosomal dominant LGMD as LGMD1A, 1B, 1C, and so forth, and autosomal recessive LGMD as LGMD-2A, 2B, 2C, and so forth.26 The current status of this classification is shown in Table 572-2. Mutations within the same gene may result in different phenotypes, sometimes not consistent with the strict definition of LGMD; for example, LGMD2B and Miyoshi distal myopathy are caused by dysferlin gene mutations, whereas mutations in the gene encoding lamin A/C may result in the phenotypes of autosomal dominant Emery-Dreifuss muscular dystrophy, LGMD1B, or cardiomyopathy with conduction system disease. Sarcoglycanopathies are early-onset autosomal recessive LGMDs caused by mutations in α, β, γ, and δ sarcoglycans, which are members of the dystrophin-associated glyco-protein complex.
Table 572-2. Limb-Girdle Muscular Dystrophies
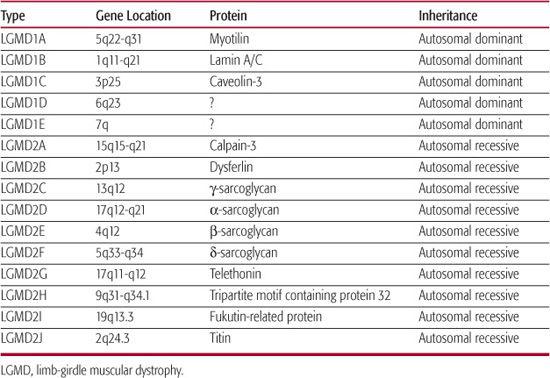
The age of onset of LGMD varies from early childhood to adulthood, but typically the onset is not congenital. In some cases, weakness may be noted early, leading to significant disability during childhood; in others, the weakness may not be apparent until early in adult life. The course is usually slowly progressive, but may be rapid in a few cases. The weakness affect the shoulder girdle (scapulohumeral type) or the pelvic girdle (pelvifemoral type) or both.
Serum creatine kinase levels are usually modestly elevated, but can be very high in sarcoglycanopathies, dysferlinopathy, and caveolinopathy. A muscle biopsy results reveal dystrophic changes. Prior to performing a muscle biopsy, DNA testing for calpain 3, sarcoglycans (if available), and fukutin-related protein (LGMD2I), as well as protein testing for dysferlin in blood, are suggested.27 In all patients with a Duchenne muscular dystrophy/Becker muscular dystrophy phenotype and no detectable dystrophin gene mutations, genetic testing for limb-girdle muscular dystrophy 2I is indicated. If genetic and protein tests are uninformative or unavailable, the next appropriate diagnostic procedure is a muscle biopsy. Immunohistochemistry with antibodies against α, β,γ, and δ sarcoglycans; dystrophin; dystroglycans; and merosin may offer a means for a specific biopsy diagnosis (eg, α-sarcoglycanopathy), but not always.
Treatment is supportive and is aimed at the prevention of contractures, as substantial disability may result from them. Therefore, a passive stretching physical therapy program is instituted early. Later in the course of the disease, cardiorespiratory monitoring is indicated.
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
The classical form of facioscapulohumeral muscular dystrophy (FSHD) is inherited in an autosomal dominant fashion and has been mapped to chromosome 4q35. In this region, deletion of an integral number of tandemly arrayed 3.3-kb repeat units (called D4Z4) has a causal relationship to FSHD.28 The general population exhibits a number of repeat units varying from 11 to more than 100; patients with FSHD exhibit deletion of an integral number of these units and an observed allele of 1 to 10 residual units.
It is hypothesized that D4Z4 contraction leads to the inappropriate overexpression of 1 or more disease genes. Hypomethylation of D4Z4 may mediate this effect. The overexpression of genes in the area may be responsible for the phenotype, but that is not certain.29
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Although FSHD is usually slowly progressive, it can be extremely variable in its severity and even the age of onset. The infantile form of FSHD has a very early onset (usually within the first few years of life) and is rapidly progressive, usually with wheelchair confinement by the age of 9 to 10 years. There is profound facial weakness, inability to close the eyes in sleep, inability to smile and to show any evidence of facial expression. The weakness rapidly involves the shoulder and hip girdles, with lumbar lordosis, pronounced forward pelvic tilt, and hyperextension of the knees and the head upon walking. Marked weakness of the wrist extensors may result in a wrist drop. The infantile variety of FSHD is often sporadic. Patients with infantile FSHD and a small number of chromosome 4q35 repeats often have associated mental retardation, epilepsy, and severe sensorineural hearing loss.
In the classical form of FSHD, the onset is usually in the second or third decade, and the progression is slow, with almost normal life span. The facial muscles are involved initially, with inability to close the eyes tightly, smile, or whistle. The facial weakness, however, can be mild early on and may remain mild for many years. The muscles of the shoulders and upper arms are also involved with marked atrophy of the biceps and triceps, but relative preservation of the deltoid muscles. There is significant scapular winging. Exudative telangiectasia of the retina (Coats syndrome) with an associated sensorineural hearing loss occurs in both infantile and classical FSHD cases. Cardiac involvement has been documented at various rates (4–60%) in a published series regarding FSHD patients.
 DIAGNOSIS
DIAGNOSIS
Serum creatine kinase levels are only mildly elevated, and rarely elevated in patients presymptomatically. A blood DNA test is available for FSHD; most patients with classic FSHD carry 1 to 10 residual repeat units within the subtelo-mere of chromosome 4q (4q35). This diagnostic test is positive in 95% to 98% of typical facioscapulohumeral muscular dystrophy cases, but the sensitivity of the genetic test for atypical cases remains uncertain. In typical cases, we see no value in performing a muscle biopsy.
 TREATMENT
TREATMENT
Treatment of facioscapulohumeral muscular dystrophy (FSHD) is primarily supportive. There is need to examine the eyes for evidence of exudative telangiectasia (Coats syndrome),30 which is usually treatable with photocoagulation of the abnormal vessels to prevent retinal detachment.
CONGENITAL MUSCULAR DYSTROPHY
Infants with hypotonia and weakness at birth in whom muscle biopsies show changes consistent with muscular dystrophy are described as having congenital muscular dystrophy (CMD). Contractures of 2 or more joints (arthrogryposis) are also commonly present in the newborn period.
Table 572-3. Genetic Loci for Congenital Muscular Dystrophy Identified to Date
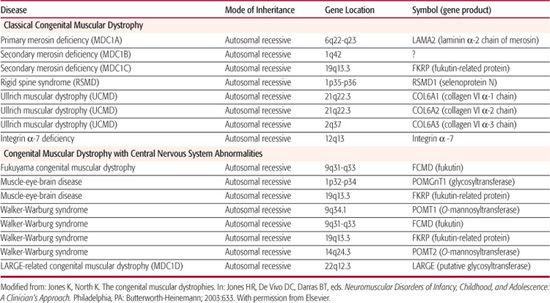
Congenital muscular dystrophy includes a number of genetically determined conditions in which muscular dystrophy is evident at birth. Serum creatine kinase concentration is usually elevated. Abnormal characteristics found as a result of muscle biopsy include extensive fibrosis, degeneration and regeneration of muscle fibers, and proliferation of fatty and connective tissue. In most patients, the clinical course progresses very slowly, although in some cases it is static. Actual improvement has been observed in a few cases.
The presence or absence of structural central nervous system abnormalities, detected by neuroimaging or at autopsy, forms the basis of CMD classification. Occidental, or classical, CMD, in which such structural changes are absent, is thereby distinguished from Fukuyama muscular dystrophy, Walker-Warburg syndrome, and muscle-eye-brain disease. Classical CMD has been further classified based on identification of mutations within the laminin α-2 chain gene (merosin) into merosin-negative and merosin-positive subgroups. The chromosomal location and respective genes identified for these disorders appear in Table 572-3.
 CLINICAL PRESENTATION AND GENETICS
CLINICAL PRESENTATION AND GENETICS
A severe classical form of CMD which is merosin-deficient, has been described in children of European ancestry combined with high creatine kinase levels and demyelination of the cerebral hemispheres without, in most cases, structural central nervous system anomalies. The associated mutated gene was mapped to chromosome 6q22-23 and identified as encoding merosin, which is the -2 chain of laminin (LAM2) and a component of the dystrophin-associated protein (DAP) complex (Fig. 572-2).31
Immunohistochemistry with antimerosin antibodies performed on muscle biopsy specimens from these patients shows a diminished or absent staining for the protein. Merosin-positive patients without structural brain abnormalities usually have a milder phenotype and are clinically and genetically heterogeneous.
Fukuyama CMD, one of the most common autosomal recessive disorders in Japan (0.7 to 1.2 per 10,000 births), is characterized by hypotonia, generalized weakness, severe developmental delay, seizures, microcephaly, and elevated serum creatine kinase levels. EEG shows epileptiform activity. Cerebral CT or MRI show cortical dysgenesis with pachygyria and polymicrogyria in the temporal and occipital regions. Patients may have simple myopia, but no structural changes to the eye.32
The genetic locus for Fukuyama CMD has been mapped to chromosome 9q31-33 and the mutated gene identified. The respective protein, fukutin, is secreted outside the cell and may be a component of the extracellular matrix reinforcing muscle membranes. Pathologic studies of the brain have suggested that fukutin is a constituent of the basement membrane.
CMD associated with ocular dysplasia, hydrocephalus, and cerebral malformations is termed cerebro-ocular dysplasia or Walker-Warburg syndrome.33 Ocular abnormalities include cataracts, optic nerve hypoplasia, corneal clouding, and retinal dysplasia or detachment. Serum creatine kinase concentration is mildly to moderately elevated, and EMG shows myopathic changes. Brain MRI shows a number of abnormalities: hypodense white matter, hypoplastic cerebellum and pons, absent vermis and corpus callosum, fused hemispheres, and ventricular dilatation with or without hydrocephalus; the pattern of abnormal cortical development, known as type II lissencephaly or cobblestone-type brain malformation, is present. Dandy-Walker cyst, sometimes with posterior encephaloceles, is also associated with this disorder. Patients with Walker-Warburg syndrome have a median survival of only 4 months.
The gene for Walker-Warburg syndrome was mapped to chromosome 9q34.1, and a subset of Walker-Warburg syndrome patients were found to have mutations in the O-mannosyltransferase POMT1 gene.34 These mutations seem to result in defective glycosylation of α-dystroglycan, which is a component of the DAP complex (Fig. 572-2). Muscle biopsy immunohistochemistry shows deficient α-dystroglycan and partial staining for merosin.
A milder phenotype than Walker-Warburg syndrome, muscle-eye-brain (MEB) disease is especially prevalent in Finland. Its typical presentation is that of hypotonia, severe progressive myopia from infancy, and developmental delay.35 With advancing age, patients develop pale retina, low or flat electroretinogram, and visual failure related to retinal degeneration. Patients commonly experience seizures and often display severe cognitive impairment. Most patients experience a decline in motor function around age 5 years, at which time they develop contractures and spasticity.
Serum creatine phosphokinase levels are elevated in MEB disease. Brain MRI in MEB disease shows cobblestone lissencephaly less severe than that seen in Walker-Warburg syndrome and also shows a characteristically flat brain stem. Immunohistochemistry on muscle biopsy in MEB disease shows normal dystrophin as well as other dystrophin-associated proteins, with the exceptions of deficient α-dystroglycan and diminished but present merosin.
The genetic locus for MEB disease has been mapped to chromosome 1p32-p34 and the disease results from mutations of the POMGNT1 gene. In MEB disease as well as in other congenital muscular dystrophies, selective deficiency and hypoglycosylation of -dystroglycan may play an etiologic role.
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Congenital muscular dystrophy (CMD) presents in the newborn period as a floppy baby, often with arthrogryposis. Serum creatine kinase levels are variably elevated in CMD. At this time, commercial DNA tests are not readily available for merosin, collagen VI, fukutin, POMT1, or POMGnT1 mutations; therefore, diagnosis is based on findings of widespread dystrophic changes in muscle biopsy specimens and on muscle immunohistochemical examination with anti-merosin, anti-collagen VI, and anti-α-dystroglycan antibodies, which usually reveal an absence of the respective protein in the sarcolemma of the muscle fibers. Brain or ocular abnormalities should be excluded by MRI and eye examination.
No definitive treatment is available for these disorders.
OTHER MYOPATHIES
CONGENITAL MYOPATHIES
Shy and Magee introduced the term congenital myopathy to describe central core disease and any myopathy present at birth excluding muscular dystrophy.36 Unfortunately, inclusion of conditions such as nemaline and centronuclear myopathies, which may be progressive and in some cases lethal, blurred the clinical distinction from the muscular dystrophies. These conditions are, however, distinct at a pathologic level. In congenital muscular dystrophies the muscle biopsy findings are dystrophic and nonspecific, whereas in congenital myopathies there are distinct myopathologic features without significant fibrosis, muscle fiber degeneration, or replacement with adipose tissue. In congenital myopathies, the specificity of distinguishing pathologic features has declined recently with the inclusion of conditions with similar but not identical histologic features, such as multicore or minicore disease. Congenital myopathies are inherited as autosomal dominant, autosomal recessive, or X-linked recessive traits.
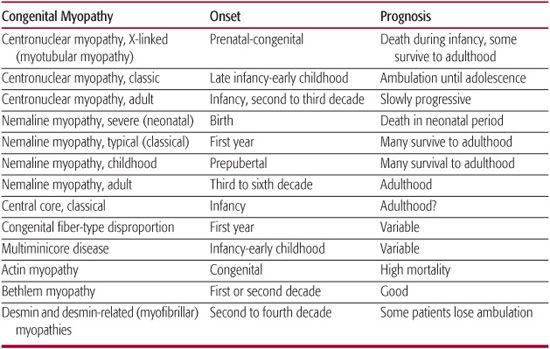
Hypotonia and weakness are the major clinical features. However, other characteristic features of congenital myopathy, such as scoliosis, ptosis, and ophthalmoplegia, may not be apparent at birth. Therefore, despite the frequent history of infantile hypotonia, diagnosis may be delayed until gross-motor developmental delay and associated weakness develop in late infancy or early childhood. In the congenital myopathies, the EMG may be normal or myopathic with small polyphasic motor unit potentials, normal nerve conduction studies, normal repetitive nerve stimulation, and absence of abnormal spontaneous activity. Serum creatine phosphokinase levels are usually normal or slightly elevated. This is a helpful distinction from congenital muscular dystrophies, in which creatine phosphokinase levels are usually moderately to markedly elevated. The diagnosis of congenital myopathies is heavily dependent on the results of the muscle biopsy, which reveals the characteristic features for which the disorder has been named. Table 572-4 summarizes the onset and prognosis of congenital myopathies.
Table 572-4. Congenital Myopathies
METABOLIC MYOPATHIES
Metabolic myopathies are rare, genetically determined conditions related to defects in glycolysis, glycogen, lipid, and mitochondrial metabolism. Classic examples are acid maltase deficiency (Pompe disease), cytochrome C oxidase deficiency, carnitine deficiency, carnitine transport enzyme defects, and fatty acid oxidation defects. Patients may present with hypotonia and static weakness, but quite often present with exercise intolerance, cardiomyopathy, hypoglycemia, and/or other systemic manifestations. Metabolic myopathies are discussed in Section 11.
INFLAMMATORY MYOPATHIES
Inflammatory myopathies are discussed in Sections 15 and 16.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


