Myeloid Malignancies
Soheil Meshinchi, Robert J. Arceci, and Howard J. Weinstein
Acute myeloid leukemia (AML) accounts for about 20% of cases of acute leukemia in children and 80% of cases of acute leukemia among adults. Further, although AML is significantly less common than acute lymphoblastic leukemia (ALL) in childhood, the survival for children with AML is current between 50% and 60% compared to nearly 85% of children with ALL. In addition, the treatment for children with AML remains particularly toxic and includes multiple, near myeloablative courses of treatment with chemotherapeutic agents and often hematopoietic stem cell transplantation (HSCT). As chemotherapeutic regimens have achieved higher cure rates in selected patients with good prognostic characteristics, HSCT is currently recommended primarily for patients with very high-risk disease characteristics or those who relapse and achieve a second remission. Insights into stem cell physiology and the molecular basis of AML have demonstrated some of the fundamental molecular changes driving the behavior of the leukemia, revealed their extensive heterogeneity, and have begun to provide new therapeutic targets and strategies.
The chronic myeloid forms of leukemia are extremely rare in children. These myeloproliferative syndromes most commonly include the adult type of Philadelphia-chromosome-positive (Ph+), chronic myelogenous leukemia (CML), and juvenile myelomonocytic leukemia (JMML). The clinical course, biologic characteristics, and molecular pathogenesis of CML and JMML are quite different. Until recently, allogeneic bone marrow transplant (BMT) from either a related or an unrelated donor was the management of choice for children with Ph + CML but the kinase inhibitor imatinib has changed the treatment paradigm for that disease in both children and adults, although hematopoietic stem cell transplantation (HSCT) is still the only know curative therapy for CML. An allogeneic HSCT remains the only know curative option for children with JMML.
ACUTE MYELOID LEUKEMIA
 EPIDEMIOLOGY
EPIDEMIOLOGY
The annual incidence of acute myeloid leukemia (AML) in children remains constant, with the exception of a slight peak in infants and during adolescence. After age 20 years, the incidence of AML slowly increases with age. Infants with congenital leukemia are more likely to have AML than acute lymphoblastic leukemia (ALL). Although the incidence of AML in children in the United States is approximately 7 cases per million children per year or approximately 600 new cases per year, there is a slightly higher incidence in Hispanic children of 9 per million per year. The incidence also appears slightly higher in Japan, Australia, and Zimbabwe. Of note, the incidence of AML has been increasing slightly although steadily.
The cause of AML is unknown, and most children have no known predisposing factors. Known risk factors include exposure to high-dose ionizing radiation, previous chemotherapy (especially with alkylating agents and epipodophyllotoxins), Down syndrome, congenital bone marrow failure syndromes (Diamond-Blackfan anemia and Kostmann agranulocytosis; see Chapter 430), chromosome fragility and impaired DNA repair mechanisms (such as Fanconi anemia), and inherited disorders, such as neurofibromatosis type I (NF1), which is due to mutations in neurofibromin, a RAS-directed GTPase (see Chapter 182). Children with NF1 are at increased risk of malignant disease, including myelodysplastic and myeloproliferative syndromes. The increased concordance of leukemia in identical twins (approximately 15%) appears to result from transplacental transfer of a single leukemic clone rather than from a genetic predisposition and approaches 100% for infant leukemia.
Children with Down syndrome have a greater than 15-fold increased risk of leukemia compared to infants without Down syndrome. During the first three years of life, acute myeloid leukemia (AML), especially the megakaryoblastic subtype, predominates, but thereafter the ratio of acute lymphoblastic leukemia (ALL) to AML follows the usual childhood distribution. Besides being at risk for acute leukemia, children with Down syndrome or trisomy 21 mosaicism are at risk of transient myeloproliferative disorder (TMD). This syndrome is usually diagnosed during the first several days to weeks after birth and cannot be reliably differentiated from congenital AML. Infants with TMD often have elevated leukocyte counts (> 50,000/μL) with circulating blasts, hepatosplenomegaly, effusions, and may have hydrops. Bone marrow aspirations from these children usually have a lower blast percentage compared to peripheral blood. Unlike congenital AML, TMD usually resolves spontaneously within several weeks to months in about 80% of cases without cytotoxic therapy. However, 5% to 10% of neonates with TMD die from hepatic failure or multiorgan failure. In some of these patients, hepatic fibrosis has been associated with megakaryoblast infiltration of the liver. The blasts from infants with TMD have been shown to be clonal, have cell surface antigens characteristic of megakaryo-blasts, and have mutations in the GATA-1 gene, an erythroid/megakaryocytic restricted transcription factor. Recent data indicate that up to 30% of neonates who have spontaneous regression of TMD will develop AML before age 3 years. It is usually of the megakaryoblastic subtype and the blasts have been shown to harbor the same GATA-1 mutation as found in the transient blast population of TMD. Interestingly, these children respond well to chemotherapy and have about an 80% likelihood of overall survival. At present, the only neonates with TMD who are recommended to be treated include those with hepatic manifestations or very high white blood cell counts at diagnosis (> 50,000/μl). Treatment for these neonates usually includes low doses of cytosine arabinoside.
The risk of secondary AML among children and adults previously treated with alkylating agents and topoisomerase-II inhibitors, especially epipodophyllotoxins, is well established. Leukemia associated with use of an alkylating agent occurs within 4 to 10 years after initial therapy, is usually associated with abnormalities of chromosomes 5 and 7, and carries a grave prognosis. Leukemia associated with use of an epipodophyllotoxin (etoposide and teniposide) has a shorter latency (2–4 years) and usually is of the myelomonocytic or monocytic subtype; characteristically, translocations involving chromosome 11q23 with rearrangement of the MLL gene are noted. Children with the latter forms of leukemia often achieve complete remission with chemotherapy but invariably relapse and die unless they are treated by means of bone marrow transplant.
 CLONALITY AND PATHOGENESIS
CLONALITY AND PATHOGENESIS
Acute myeloid leukemia (AML) is a clonal disorder that is the consequence of acquired molecular alterations in hematopoietic progenitor cells that cause differentiation arrest and confer proliferative growth advantage to the affected clone. Early studies using X-linked polymorphism and more recent molecular techniques have demonstrated clonal origin of AML, showing that leukemic cells share common genetic composition and are believed to have been derived from a common ancestral cell.  One major contributor to AML pathogenesis is acquisition of cyto-genetic abnormalities, including balanced or unbalanced chromosomal translocation, deletion or duplication of a region, or the entire gene. In childhood AML, approximately 80% of the patients have one of more than 300 identified cytogenetic abnormalities ranging in prevalence from less than 1% to greater than 15%. In addition, somatically acquired mutations in genes involved in AML pathogenesis have been identified that cooperate with specific cytogenetic alterations to cause AML phenotype.
One major contributor to AML pathogenesis is acquisition of cyto-genetic abnormalities, including balanced or unbalanced chromosomal translocation, deletion or duplication of a region, or the entire gene. In childhood AML, approximately 80% of the patients have one of more than 300 identified cytogenetic abnormalities ranging in prevalence from less than 1% to greater than 15%. In addition, somatically acquired mutations in genes involved in AML pathogenesis have been identified that cooperate with specific cytogenetic alterations to cause AML phenotype.
During a morphologic remission, the clone is no longer detectable except with molecular methods. During hematologic relapse, the original clone reappears. The transforming event in acute myeloid leukemia (AML) could occur at any point in hematopoiesis from the pluripo-tent stem cell to a committed precursor, such as the myeloblast or erythroblast. Both animal and human data, however, provide evidence that the leukemic stem cell in AML is a primitive hematopoietic stem cell in most instances.
 GENETIC FEATURES
GENETIC FEATURES 
Cytogenetic abnormalities are identified in nearly 80% of childhood AML, many of which are unique to AML. Nearly 300 recurrent cyto-genetic abnormalities including translocations, deletions, or duplications have been identified. The most common chromosomal abnormalities in children and young adults with AML include inv16, t(15;17), (8;21), and chromosome 11q23 abnormalities (Fig. 450-1), which account for nearly half of the AML cases, and occur at a higher rate in pediatric populations compared to adults. Other cytogenetic abnormalities, including monosomy 5 or deletion 5q, monosomy 7, and trisomy 8, are less common in children and have a higher prevalence in adults. The molecular events associated with many of the structural chromosomal changes have now been elucidated. 
 CLASSIFICATION
CLASSIFICATION
Precise diagnosis and classification is essential to successful management and biologic investigation of childhood leukemia. In 1976, the French-American-British (FAB) Cooperative Group proposed a classification system based primarily on morphology and cytochemical features of the blasts and required at least 30% bone marrow blasts for the diagnosis of acute myeloid leukemia (AML). Although FAB classification provided a valuable tool for general classification of AML, it lacked the ability to predict accurately cytogenetic subclasses, and, in general, did not provide reliable prognostic information.
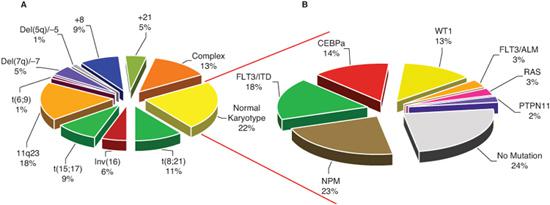
FIGURE 450-1. Molecular alterations in childhood acute myeloid leukemia (AML): (A) prevalence of cytogenetic abnormalities in childhood AML and (B) prevalence of mutations in patients with normal karyotype.
In 2002, the World Health Organization (WHO) proposed a new classification system that incorporated diagnostic cytogenetic information, which more reliably correlated with outcome into AML classification.  More recently, WHO expanded the number of cytogenetic abnormalities linked to AML classification, and for the first time included specific gene mutations (FLT3, CEBPA, and NPM mutations) in its classification system (Table 450-1). Such a genetically based classification system links AML class with outcome and provides significant biologic and prognostic information. With new emerging technologies aimed at genetic, epigenetic, proteomic, and immunophenotypic classification, AML classification will continue to evolve and provide informative, prognostic, and biologic guidelines to clinicians and researchers.
More recently, WHO expanded the number of cytogenetic abnormalities linked to AML classification, and for the first time included specific gene mutations (FLT3, CEBPA, and NPM mutations) in its classification system (Table 450-1). Such a genetically based classification system links AML class with outcome and provides significant biologic and prognostic information. With new emerging technologies aimed at genetic, epigenetic, proteomic, and immunophenotypic classification, AML classification will continue to evolve and provide informative, prognostic, and biologic guidelines to clinicians and researchers.
 CLINICAL AND LABORATORY FEATURES
CLINICAL AND LABORATORY FEATURES
The initial signs and symptoms for most children with acute myeloid leukemia (AML) include anemia, thrombocytopenia, and neutropenia caused by bone marrow infiltration with leukemic blasts and decreased production of normal cells. Patients commonly present with pallor, fatigue, epistaxis, gum bleeding, petechiae, or purpura, as well as fever or infection that has not responded to antibiotic therapy. Children with AML may have bone or joint pain, but these symptoms occur more often in children with acute lymphoblastic leukemia (ALL). Bulky peripheral lymphadenopathy is not a common finding, and massive hepatosplenomegaly is rare with AML except among infants. Extramedullary leukemia can present as gingival hyperplasia, central nervous system (CNS) leukemia (headache, cranial nerve palsy), and skin nodules. Neonates and infants with AML frequently have leukemia cutis characterized by a papular or nodular rash that is salmon or bluish to slate gray in color. Clinical findings of CNS leukemia at diagnosis are rare. They include signs of increased intracranial pressure or cranial nerve palsy, seventh nerve palsy being the most common. Fewer than 5% of patients with AML have myeloblastomas (also known as granulocytic sarcoma or chloroma) at diagnosis or during the course of the illness. These are solid tumors of blasts and immature myeloid cells that typically occur in the bones and soft tissues of the head and neck (often involving the orbits), intracranial or epidural sites.
Peripheral blood counts at diagnosis in children with AML can be quite varied. The leukocyte count ranges from less than 1000/μL to more than 500,000/μL. Approximately 15% to 20% of children have an initial leukocyte count greater than 100,000/μL. Higher leukocyte counts are associated with the FAB, M4, and M5 subtypes, whereas lower leukocyte counts (< 5000/μL) are commonly seen in acute promyelogonous or M3 leukemia (acute promyelocytic leukemia [APL]). Most patients have a normocytic anemia (median hemoglobin concentration of 7 g/dL in one series), and approximately 50% of patients have platelet counts less than 50,000/μL. Disseminated intravascular coagulation is extremely common among almost all patients with APL and some infants with monocytic leukemia.
The characteristic bone marrow findings include hypercellularity with more than 20% blasts (usually 70–90% blasts). A bone marrow biopsy infrequently shows myelofibrosis (except for megakaryoblastic) and occasional multilineage dysplasia.
In most cases of AML, the diagnosis is straightforward after examination of the peripheral blood sample and a bone marrow aspirate. Other conditions that can cause diagnostic difficulty include the myeloproliferative disorders such as juvenile myelomonoctic leukemia, myelodysplastic syndromes, sepsis that causes a leukemoid reaction, or neutropenia caused by maturation arrest in granulocytic-monocytic precursors. In the presence of sepsis, the bone marrow findings may suggest acute promyelocytic leukemia because of a promyelocyte arrest with toxic granulation. However, normal granulocytic maturation ensues within a few days with resolution of the infection. As previously discussed, acute myeloid leukemia among neonates with Down syndrome is difficult, if not impossible, to differentiate from transient myeloproliferative disorder (TMD).
 MANAGEMENT OF NEWLY DIAGNOSED ACUTE MYELOID LEUKEMIA
MANAGEMENT OF NEWLY DIAGNOSED ACUTE MYELOID LEUKEMIA
Substantial improvement in survival rate from less than 10% to approximately 50% of children with acute myeloid leukemia (AML) has occurred during the past 30 years. The improvement is the result of a higher percentage of children entering complete remission, a decrease in relapse rate because of more effective postremission strategies, including allogeneic hematpoietic stem cell transplantation (HSCT), and improvements in supportive care. All children with AML should be referred to pediatric oncology centers and treated on clinical trials.
Induction of Remission
The most widely used remission-induction regimen includes treatment with an anthracycline (usually daunorubicin) and cytarabine arabino-side with or without thioguanine or etoposide. With these regimens, 85% to 95% of children with acute myeloid leukemia (AML) enter complete remission after receiving 1 to 2 cycles of induction chemotherapy. Because the remission-induction phase of therapy is associated with prolonged cytopenias (3–5 weeks), 2% to 5% of patients may die of infectious or hemorrhagic complications before completing the induction phase. Deaths during the first several days after diagnosis are rare and often are caused by leukostasis or disseminated intravascular coagulation. Leukostasis, or plugging of blasts in vessels, is associated with elevated peripheral blast counts (more than 100,000/μL) and can cause hemorrhagic infarction of the brain or other organs. A greatly elevated leukocyte count is a medical emergency, and measures should immediately be taken to decrease the leukocyte count with chemotherapy (eg, hydroxyurea), exchange transfusion, or leukopheresis if the patient has symptoms, such as hypoxemia or mental status changes. Intensifying the doses or timing of chemotherapy during the remission-induction phase of treatment has not increased the percentage of children achieving complete remission but has resulted in a decrease in relapse rates and improvement in overall survival rates.
Table 450-1. WHO Classification of AML



