Mary Brennan, Helen Foster, Flora McErlane, Rajib Lodh, Sharmila Jandial • Understand the embryology and anatomy of the musculoskeletal system • Understand the histology and physiology of normal muscle and how structure relates to function • Understand the pathophysiological changes which occur in muscle and joint disorders • Know the commoner genetic and environmental factors in the aetiology of musculoskeletal disorders • Know the investigations used in the diagnosis of musculoskeletal disorders The embryological development of the musculoskeletal (MSK) system is complex with many components (bones, skeletal muscles, connective tissues (cartilage, ligaments and tendons), blood vessels and nerves). Development requires differentiation into these specific cell types and coordination to produce an integrated and functional system. This complexity results in problems that can and do arise and MSK developmental abnormalities are one of the largest groups of congenital conditions. The MSK system mainly develops from the mesodermal germ layer with some neural crest contribution. Mesoderm is subdivided by position into three parts: the paraxial, intermediate and lateral mesoderm. In the 3rd week of gestation, the paraxial mesoderm forms ‘little balls’ (somites), which are paired each side of the neural groove. These somites differentiate differently in different regions. The sclerotome eventually splits segmentally giving rise to the vertebral column, and the dermomyotome develops into dermal and muscle components. Lateral mesoderm and dermomyotome cells migrate to the limb field and proliferate to create the limb buds. Mesodermal cells give rise to mesenchyme, which is loosely organized connective tissue. Mesenchymal cells are pluripotent and differentiate into many different cell types, including those that give rise to bones and cartilage. Whilst the cranial bones of the skull and clavicle form from direct ossification from mesenchyme, the bones of limb and girdle form from ossification of a cartilaginous precursor of the long bones of the skeleton (a process known as endochondrial ossification). Condensation of lateral plate mesenchyme occurs in a rod-like structure along the axis of the limb bud. Cartilage cells (chondrocytes) respond to growth factors and differentiate (chondrification). These cells secrete proteoglycans and collagen to form cartilage. Initially there is deposition of cartilage around the entire limb condensations but then further chondrification is limited to future bone sites, sparing the interzone regions, resulting in the site of future joints. For the joints of the long bones, mesenchymal cells at the interzones of long bones differentiate into multiple fibroblastic connective tissue layers. These then differentiate further to provide the articular cartilage at either end of the joint, and connective tissue in the middle forms the internal structures of the joint – synovial tissue, menisci and ligaments. Vacuoles form within the connective tissue which then becomes the joint cavity. The mesenchymal sheath becomes the joint capsule (Fig. 27.1). Fibrous joints, or immobile joints which connect bones (e.g. in the skull, pelvis) are also developed from interzones, which differentiate into a single layer of fibrous connective tissue. Following chondrification, ossification occurs from the primary ossification centre. In response to growth factors, mesenchymal cells in this area differentiate into bone cells (osteoblasts). These cells secrete the calcium matrix of mineralized bone. Bone-resorbing osteoclasts also appear. These enable remodeling of growing bone, a process which continues throughout development into adult life. The limb musculature develops from two condensations of somitic mesoderm that in the 5th week invades the limb bud, one ventrally, the other dorsally. Ventral somatic mesoderm gives rise to mainly flexors, pronators and adductor muscles whilst dorsal somatic mesoderm dorsally gives rise to mainly extensors, supinators and abductor muscles. Cells in these condensations differentiate into myoblasts (muscle cell precursors). Myoblasts fuse together to form syncytia. The innervation of the limb muscles develops from branches that develop from spinal nerve axons in a multistep process. Branches of the ventral spinal nerve innervate ventral muscles, and branches of the dorsal spinal nerve innervate dorsal muscles. Limb defects can be grouped into three categories: Limb defects can be broadly attributed to the following processes: • Development arrest (failure to form) • Focal defects, e.g. amniotic fluid band syndrome • General skeletal abnormalities, e.g. osteogenesis imperfecta (see below) Most limb defects have multifactorial aetiologies, from a combination of environmental and genetic influences, but either may predominate. Examples of conditions where genetic (familial) aetiologies predominate include most causes of polydactyly or ectrodactyly (absence of fingers or toes). Polydactyly is a relatively common finding in newborns, affecting 1 in 500 newborns. Over 100 genes have been described but most cases result from abnormalities in a single gene. Postaxial hand polydactyly is a common isolated disorder in Black African children, especially males, and autosomal dominant transmission is suspected. In contrast, postaxial polydactyly seen in white children is usually syndromic and associated with an autosomal recessive transmission. The cause of developmental dysplasia of the hip (DDH) is not clear but it is usually multifactorial. Known risk factors include female gender, family history, oligohydramnios, breech position, and the presence of other congenital abnormalities, e.g. neuromuscular disorders. In the normal development of the hip joint, the head of the femur is smooth and rounded, and the acetabulum is cup-shaped. However, in DDH there may be abnormalities of the shape of the head of the femur, the shape of the acetabulum or the surrounding structures. This means that the acetabulum and femur may not be in close contact. Depending on the degree of abnormality, the hip may be subluxed or dislocated. Talipes equinovarus is a complex abnormality affecting 1 in 1000 live births. The entire foot is inverted and supinated, the forefoot adducted and the heel is rotated inwards and in plantar flexion. The affected foot is shorter and the calf muscles thinner than normal. The position of the foot is fixed, cannot be corrected completely and is often bilateral. It is more common in males (2 : 1) and can be familial but is usually idiopathic. However, it may also be secondary to oligohydramnios during pregnancy, a feature of a malformation syndrome or of a neuromuscular disorder such as spina bifida. The Ponseti method for correction is a manipulative technique involving serial casting and usually avoids the need for invasive surgery. Talipes equinovarus needs to be differentiated from positional talipes, where the foot is of normal size, the deformity is mild and can be corrected to the neutral position with passive manipulation, and the rare congenital vertical talus, where the foot is stiff and rocker-bottom in shape. Many of these infants have other malformations. Many drugs which might be taken during pregnancy, particularly if taken between 4–8 weeks’ gestation, can result in limb defects: In utero limb growth requires physical space. Constricted uterine environment or intrauterine compression, e.g. bicornate uterus or multiple pregnancy, can result in deformity of a limb. Occasionally, fibrous amniotic tissue detaches and wraps around the developing limb, forming an amniotic band. This physical stricture can prevent growth or development of a limb or part of a limb. Amniotic band syndrome affects up to 1 in 1200 live births and is associated with increased risk of other abnormalities, including cleft lip and palate and talipes equinovarus. Congenital structural defects of the spine may occur secondary to failure of vertebrae formation, separation or fusion, e.g. abnormal induction of the sclerotome and the neural tube. Depending on the position of the abnormality, the spine may develop to the left or right (scoliosis), or convex overcurvature (kyphosis) or sway backwards (lordosis). Growth potential depends on the involvement of the vertebra growth plate and gives rise to the differences in progression and deformity. Congenital scoliosis is the most common developmental spine condition. Although the aetiology is not fully understood, failure of sclerotome segmentation results in vertebrae that are abnormally connected on one side, causing asymmetrical growth rates. There may also be disruption of the normal shape of the vertebra causing a wedge shape. Similarly, asymmetrical growth may occur. This is different to idiopathic scoliosis, which presents with either early onset (less than 5 years old) or late onset. The most common presentation is late-onset idiopathic scoliosis, mainly in girls 10–14 years of age during their pubertal growth spurt. Secondary scoliosis is common and related to other disorders that result in neuromuscular imbalance, e.g. cerebral palsy, muscular dystrophy, polio; disorders of bone or cartilage, such as neurofibromatosis, Marfan’s syndrome; or leg length discrepancy. If a child has a congenital vertebral abnormality, other systems should be investigated to exclude syndrome characteristics, e.g. VACTERL association. Arthrogryposis is the collective term for a number of conditions associated with joint contractures at birth. Factors that inhibit normal joint development or movement (extrinsic or intrinsic) can result in joint contractures. Extrinsic factors may include environmental problems, such as oligohydramnios or viral infections. Intrinsic factors include underlying genetic/molecular abnormalities leading to connective tissue, muscle or neurological abnormalities. Arthrogryposis is divided into three main categories: At birth, the diaphyses (‘shafts’) of the long bones are completely ossified, whereas the epiphyses (‘ends’) remain cartilaginous. Secondary ossification centres arise from the epiphyses, resulting in the ends gradually ossifying. Growth of the long bones relies on proliferation of chondrocytes in the epiphyseal cartilaginous plate (growth plate), which persists between the epiphysis and growing end of diaphysis (metaphysis). The epiphyses and diaphysis fuse around the age of 20 years. During childhood, if trauma occurs to the growth plate, the growth of that limb may be affected. During the ossification process, bone is invaded by numerous blood vessels, which branch from the limb vasculature. A dominant nutrient artery develops, which nourishes the bone. During life, if this nutrient vessel becomes interrupted then cellular death of bone components occurs (avascular necrosis) and if this involves the bones around a joint, the articular surface may become damaged (osteochondritis dissecans). The blood supply in certain areas is vulnerable. Osteochondroses are a group of conditions in which the primary or secondary ossification centre undergoes avascular necrosis, and there is resorption of old dead bone, with replacement of new bone tissue. Examples include: • Perthes disease – femoral head • Osgood–Schlatter disease – tibial tubercle • Freiberg disease – second metatarsal head • Thiemann disease – phalangeal epiphyses • Scheuermann disease – lumbar or midthoracic Legg–Calvé–Perthes, or Perthes disease, is one of the most common of the osteochondroses, most often seen in young boys (age range: 4–8 years; male : female ratio: 4.5 : 1). Interruption of the blood supply causes avascular necrosis of the capital femoral epiphysis of the femoral head, and is followed by revascularization and reossification over 18–36 months. Slipped capital femoral epiphysis (SCFE) results in displacement of the epiphysis of the femoral head posteroinferiorly, requiring prompt treatment in order to prevent avascular necrosis. It is most common at 10–15 years of age during the adolescent growth spurt, particularly in obese boys, and is bilateral in 20%. The cause of the osteochondroses is not clear, but they may be associated with trauma, especially at sites where vascular supply to the bone is vulnerable (e.g. femoral head) or follow a transient synovitis (e.g. Perthes disease after an irritable hip) or can be associated with metabolic endocrine abnormalities (e.g. hypothyroidism and hypogonadism in slipped capital femoral epiphysis (SCFE)). Bones consist of two cell types, osteoclasts (bone-resorbing cells) and osteoblasts (bone-forming cells). These act together to control bone growth and metabolism through the bone remodeling cycle. Each remodeling cycle takes several weeks to complete. In childhood, resorption and formation usually occur at the same rate, except when extra bone matrix is required for growth (for example, during skeletal development). In pathological situations, increases in bone resorption or decreases in bone formation may result in an overall net loss of bone (osteoporosis). Inadequate mineralization of the bone matrix (osteomalacia) can cause bone deformity if it occurs in a growing skeleton (rickets). Osteoporosis may be classified into primary or secondary causes. There are two main types of primary osteoporosis. Idiopathic juvenile osteoporosis may present pre/early puberty with metaphyseal and vertebral crush fractures. Osteogenesis imperfecta presents at any age with varying degrees of severity. The principal features include bone fragility and low bone mass. Osteogenesis imperfecta A 6-month-old was taken to the emergency department by his parents due to inconsolable crying. He was found to not be moving his right leg and an X-ray confirmed a fractured femur. X-rays revealed several other fractures in various stages of healing. His parents could not explain what may have caused them. He was referred to the safeguarding team with suspected non-accidental injury. Subsequent investigations revealed a mutation in the COL1A1 gene. Secondary osteoporosis can result from inflammatory conditions (e.g. inflammatory bowel disease, juvenile idiopathic arthritis (JIA), cystic fibrosis, prolonged use of glucocorticoids or following disuse (e.g. cerebral palsy, muscular dystrophies, immobility) and in children and young people with endocrine disturbances (e.g. thyroid disease and hypogonadal states). Most cases of osteomalacia are due to lack of vitamin D (see later in this chapter), resulting from poor sunlight exposure or dietary inadequacy. Osteomalacia is more common in darker-skinned individuals living in colder climates. Vitamin D deficiency results in myopathy and MSK aches and pains. Infants are usually miserable, and other features can include bowed limbs, metaphyseal swelling, bossed forehead and bone pain. Inherited forms of rickets include X-linked hypophosphataemic rickets, and children with McCune–Albright syndrome/polyostotic fibrous dysplasia. In this rare disorder, there is increased bone mass due to failure to resorb bone – the bones are dense but brittle. The severe autosomal recessive disorder presents with faltering growth, recurrent infection, hypocalcaemia, anaemia and thrombocytopenia (due to bone marrow failure). Prognosis is poor, but bone marrow transplantation can be curative. A less severe autosomal dominant form may present during childhood with fractures. Skeletal dysplasias are a group of clinically and genetically heterogeneous disorders causing generalized abnormalities of bone growth and/or bone modeling. Most are rare and usually present with one or more of: • Short stature (usually disproportionate). If the limbs are short, it is important to note which segment(s) are affected: – Proximal segment (shoulder to elbow, or hip to knee) – rhizomelic – Middle segment (elbow to wrist, knee to ankle) – mesomelic – Distal segment (hands, feet) – acromelic, or all segments micromelic • Recurrent fractures usually after minimal trauma Joints may be classified by their movements and tissues. Synovial joints (diarthroses) move freely, have a fibrous joint capsule and distinct joint cavity. There are several distinct subtypes including: Synovial joints are lined with a highly vascular synovial membrane. Synovial fluid is produced naturally to lubricate the joint but can be produced in excess if the synovial membrane becomes inflamed (synovitis – see juvenile idiopathic arthritis, JIA). The blood supply of the bone is an important factor in the presence of infection such as septic arthritis, as it can lead to bone destruction and usually results from haematogenous spread, especially in neonates and young children, where spread from adjacent osteomyelitis into joints can occur where the capsule inserts below the epiphyseal growth plate. Other problems may arise in joints due to disruption of supportive cartilage (e.g. meniscal problems at the knee) or ligaments. An appreciation of muscle histopathology and physiology allows the clinician to better appreciate and understand the spectrum of skeletal muscle diseases. Muscle biopsy samples may be helpful. The structure of skeletal muscles and a simplified description of how muscles contract are summarized in Figure 27.2. Muscle cells respond to electrical impulses by contracting in size and developing tension. Motor neurons stimulated by electrical impulses release acetylcholine into the neuromuscular synapse. When acetylcholine binds to the motor endplate of the muscle fibre, it results in the initiation of an action potential throughout the cell membrane of the muscle fibre (or the sarcolemma). The method by which muscle excitation (the presence of an action potential in the muscle) results in contraction of muscle fibres depends on excitation–contraction coupling. This process can be thought of as consisting of three elements: 1. Release of calcium stores from the sarcoplasmic reticulum 2. Calcium-dependent activity of mitochondria in order to produce adenosine triphosphate (ATP) At the start of exercise, when there is insufficient oxygen available for the Krebs cycle to occur effectively, ATP is generated from creatine phosphate stores in muscle fibres by the action of the enzyme creatine kinase. This store of creatine phosphate is soon depleted. During brief, vigorous exercise, pyruvate is converted to acetyl-coenzyme A (acetyl-CoA) which then enters the mitochondrial Krebs cycle. This process leads to generation of ATP (adenosine triphosphate). During prolonged exercise, free fatty acids are oxidatively converted by mitochondria to acetyl-CoA in order to enter the Krebs cycle. During anaerobic exercise, the small amount of ATP generated from the breakdown of glycogen to pyruvate is the primary source of ATP. The build-up of pyruvate leads to lactic acid formation, as there is insufficient oxygen available for the pyruvate to enter the Krebs cycle. There is a wide range of pathological processes which cause skeletal muscle disorders. Pathologies either affect the nerves (neuropathies) or the muscles directly (myopathies). Diseases that cause degeneration and destruction of muscle fibres are known collectively as muscular dystrophies. Muscle fibre development and maturation may be delayed (for example in congenital inherited myopathies) or may be affected by abnormalities in the metabolic or signaling pathways. These conditions are described in Chapter 5, Developmental problems and the child with special needs. A number of infections (e.g. influenza, Coxsackie B) can result in a muscle inflammatory response (myositis). In addition, myositis may be part of a number of systemic inflammatory conditions (e.g. juvenile dermatomyositis, systemic lupus erythematosus and systemic sclerosis/mixed connective tissue disease). Juvenile dermatomyositis A 9-year-old girl was referred with a 7-month history of gradual tiredness and weakness. She had become unable to climb the stairs and frequently complained of pains in her legs. Her mother described a persistent ‘sunburn’ rash across her face. Blood tests revealed raised creatine kinase (CK) of 600 IU/L. Muscle biopsy of her quadriceps revealed an inflammatory infiltrate, in a perivascular distribution with variation in muscle fibre size. Additionally, any condition which leads to derangement of cellular potassium, calcium or magnesium can lead to a global muscle dysfunction. This is discussed in the case history below and in Chapter 6, Paediatric emergencies and critical care, and Chapter 19, Nephrology, in more detail. Hyperkalaemic periodic paralysis A two-year-old girl presents with intermittent symptoms of profound weakness. In between these events, she is well. Each event typically lasts for 2 hours, but may last up to 12 hours and she may have several attacks in one day. The paralysis usually involves her shoulders and hips. Her facial, respiratory and bulbar muscle movements are preserved. Her parents report that these events usually occur when she is resting following a period of activity. Her father had similar episodes, although milder, and these became less prevalent after he reached the age of 30. Her serum potassium is raised during one such event. Abnormalities of the MSK system are seen in a number of genetic conditions (chromosomal, single gene and mitochondrial disorders). Commoner examples of these are summarized in Tables 27.1 and 27.2. Some features may be present at birth (e.g. hypotonia), but often become apparent as the MSK develops during childhood (e.g. hypermobility). Marfan’s syndrome A 10-year-old boy, who was the tallest in his class, was referred to ophthalmology by an optician as he had been having problems seeing clearly. He was found to have upward subluxation of the lens. Two months later, he presented to the local emergency department with acute onset of shortness of breath. His father was also tall and attended regular cardiology follow-up.
Musculoskeletal disorders
Embryology of the musculoskeletal system
Endochondrial ossification
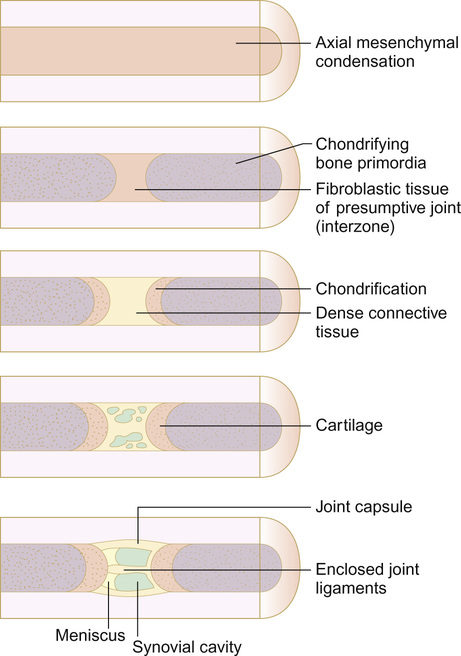
Joint formation
Primary ossification
Skeletal muscle
Congenital abnormalities
Limb defects
Developmental dysplasia of the hip
Talipes equinovarus
Environmental causes of limb defects
Drug teratogens
Amniotic tissue/constricted uterus
Spine defects
Joint defects
Skeletal development and pathology
Bones
Bone remodeling
Primary osteoporosis
 Case history
Case history
Secondary osteoporosis
Osteomalacia/rickets
Osteopetrosis (marble bone disease)
Skeletal dysplasias
Joint problems
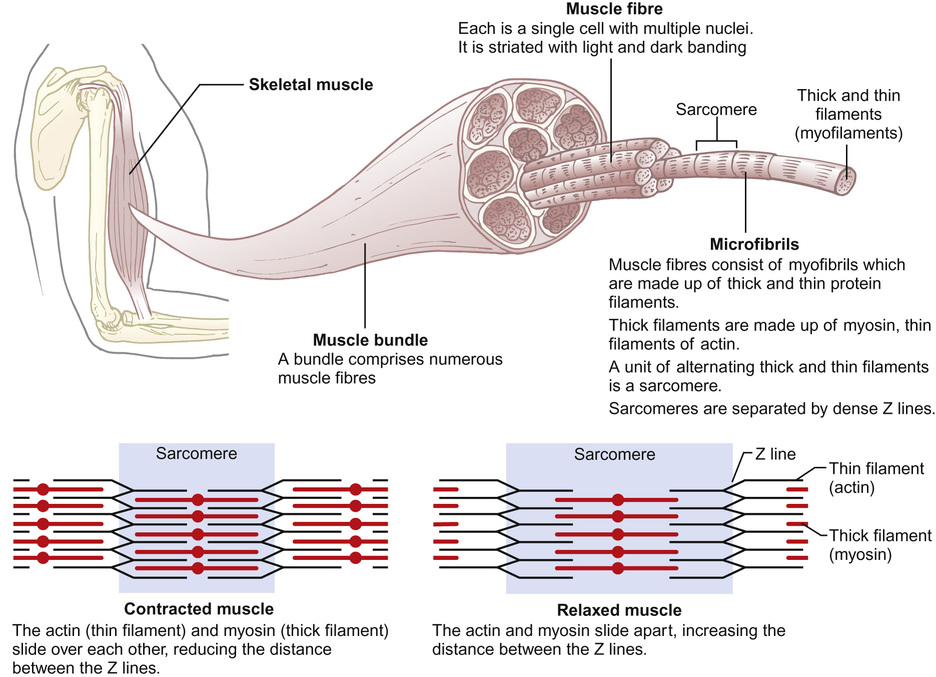
Histology and physiology of muscles
 Case history
Case history
 Case history
Case history
Genetics
 Case history
Case history