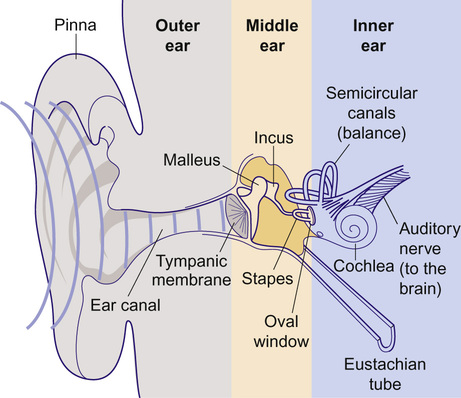
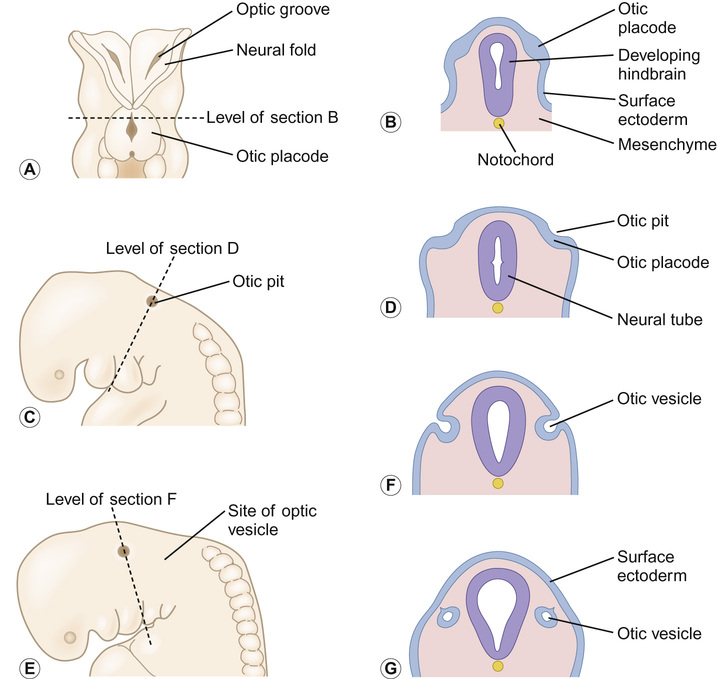
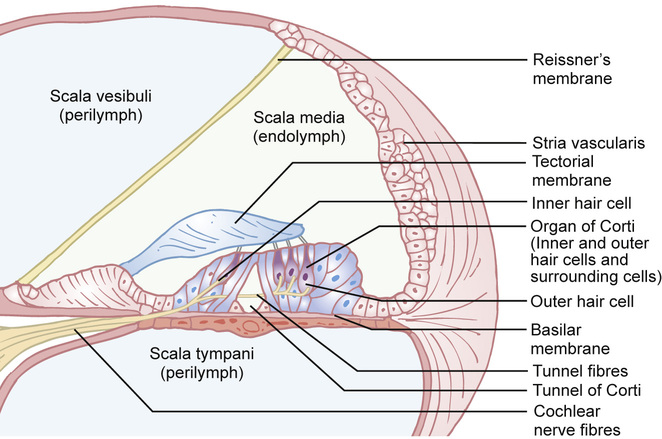
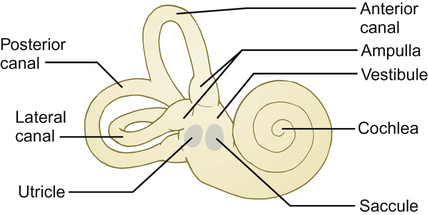
Kaukab Rajput, Maria Bitner-Glindzicz • Know the anatomy of the outer, middle and inner ear • Understand how the structure of the ear is related to function • Understand the process of hearing and how it may be impaired • Understand the process of ear development • Be aware of the genetic and environmental causes of hearing loss • Know how to investigate hearing loss • Understand about the assessment of hearing The ear is divided into the outer (external) ear, the middle ear and the inner ear. The outer ear consists of the auricle (pinna) and external auditory canal; the middle ear cavity is bordered by the tympanic membrane and contains the three ossicles; the inner ear (or labyrinth) consists of the cochlea (the organ of hearing) and the vestibular system (the organ of balance). Sound waves are collected by the pinna and channelled by the external auditory canal to the tympanic membrane, causing it to vibrate (Fig. 31.1). The shape and resonance of the pinna and external canal results in passive amplification of mid- and high-frequency sounds. Comparison of the inputs from both ears allows localization of the sound source. Children with unilateral hearing loss find it hard to localize sound or to understand speech in spatially separated background noise. The middle ear is an air-filled cavity containing the malleus, incus and stapes. It transfers sound energy from air compression waves to pressure waves in the fluids of the cochlea. Normally, the loss of energy when sound waves in air hit a fluid medium is around 99.9%, equivalent to about 30 dB, but the function of the middle ear is to offset this energy loss. The footplate of the malleus rests on the tympanic membrane, and detects vibrations from sound. This in turn is transmitted via the incus to the stapes, whose footplate rests on the oval window of the cochlea. The piston-like movement of the stapes footplate on the oval window sets up a travelling sound wave in the cochlear fluid (see below). The lever system of the ossicles and the difference in surface area of the large eardrum at one end, and the small stapes footplate at the other, means that sound is amplified more than twentyfold between the outer ear and the inner ear, offsetting the loss of energy between air and fluid. The middle ear also contains two small muscles, the stapedius muscle, innervated by the facial nerve (VII) and the tensor tympani, innervated by a branch of the trigeminal nerve (V). These contract in response to loud sounds, reducing transmission of sound to the cochlea and protecting inner ear structures from damage. This response is known as the stapedial or acoustic reflex, and involves the VIIIth nerve, the brainstem nuclei and the VIIth nerve. Clinical measurements of the stapedial reflex can help to differentiate between certain types of hearing loss, and the possible location of the lesion. Connecting the middle ear to the nasopharynx is the Eustachian tube, which regulates the air pressure in the middle ear. Most of the time this is closed (collapsed), but swallowing, yawning and positive pressure may force it open transiently. The normal cochlea is a coiled structure with two and a half turns. It is divided lengthways into three fluid-filled compartments by two membranes, the basilar membrane and Reissner’s membrane. The scala tympani is the lower compartment, the cochlear duct (scala media) is the middle one and the scala vestibule is the upper compartment (Fig. 31.2). The scala vestibuli and the scala tympani are filled with perilymph, which has a composition similar to extracellular fluid, while the cochlear duct contains endolymph, which is unique, having a high potassium and low sodium concentration. The specialized sensory hair cells, responsible for converting sound energy into electrical impulses, and their supporting cells are known as the organ of Corti. The organ of Corti runs along the entire length of the basilar membrane. The vestibular part of the inner ear (labyrinth) is responsible for balance. It is divided into two functionally separate parts: the semicircular canals and the vestibule (Fig. 31.3). It contains an internal compartment (membranous labyrinth) containing endolymph which is surrounded by perilymph and these fluids are continuous with those of the cochlea. The membranous labyrinth is encased in bone. The three semicircular canals (SCCs) include the lateral (horizontal) canal, anterior (superior) canal and a posterior (inferior) canal, at approximately right angles to each other. The anterior and posterior canals detect rotation in the vertical planes (i.e. when the head is nodding or rolling), and the lateral canal detects horizontal movements (turning the head to the left or right). At the end of each semicircular canal is a dilatation called the ampulla, containing a patch of sensory hair cells called the cristae ampullares (see Balance and the vestibular system, below). The vestibule has two parts, containing the utricle and the saccule. These both contain an area (macula) of sensory hair cells. The areas are at right angles to each other and detect gravity and linear acceleration, such as when going up in a lift (vertical), or stopping and starting in a car at traffic lights (horizontal). Together, the utricle and saccule are called the otolith organs. When the stapes footplate moves as a result of vibrations transmitted from the tympanic membrane, pressure waves in the cochlear fluid produce movement of the basilar membrane. On the basilar membrane is the organ of Corti (see Fig. 31.2), which detects this movement and converts it into electrical energy (auditory transduction). The organ of Corti consists of sensory cells (hair cells) and supporting cells. The hair cells are so called because they have hair-like, actin-filled projections on their apical surface, called stereocilia. There are three rows of outer hair cells and one row of inner hair cells. The stereocilia are arranged in rows of increasing height in a ‘W’ shape on the top of the outer hair cells, whereas the inner hair cells have a linear arrangement of stereocilia. The tallest stereocilia are embedded in an overlying gelatinous membrane, the tectorial membrane, and adjacent stereocilia of each hair cell are connected to each other by thin links, to form a hair bundle. Inward movement of the stapes in response to sound transmitted through the middle ear produces deflection of the stereocilia towards the tallest row. This increases tension in the links between the tip and the side of the adjacent taller stereocilium. This causes physical opening of ion channels at the top of the stereocilia (like the pulling open of a trap door). Ions from the potassium and calcium-rich endolymph flow into the hair cells along an electrochemical gradient, causing depolarization of the hair cells. In turn, depolarization of the hair cells triggers the opening of voltage-gated calcium channels, and further influx of calcium. This increase in calcium concentration results in exocytosis of vesicles containing neurotransmitters at the base of the hair cell, increase in firing of afferent neurons, and transmission of impulses to the brain via the cochlear nerve. However, movement of stereocilia in the opposite direction closes the channels. This causes hyperpolarization of the cell and reduction of firing. On the lateral wall of the endolymphatic (cochlear) duct is a specialized layered tissue called the stria vascularis. This produces the endolymphatic fluid, with its high potassium and low sodium concentration. The endolymph is also at a high positive potential (endocochlear potential). It is because of the electrochemical gradient between the endolymph and intracellular fluid that potassium flows into the hair cells when the channels at the top of the stereocilia are opened. Nerve impulses generated by sound are transmitted via the cochlear nerve with the vestibulocochlear (VIIIth) nerve to the cochlear nucleus, superior olive, inferior colliculus, medial geniculate body and up to the auditory cortex located in the temporal lobes. The auditory system is organized tonotopically, i.e. according to frequency (pitch) of the sound. This means that high frequencies are detected at the base of the cochlea and low frequencies at its apex, and this ‘tonotopicity’ is retained in most of the higher levels of the auditory pathways. The higher centres integrate peripheral inputs for functions such as localization and language processing. The latter is performed by association areas (primarily Broca’s and Wernicke’s areas), with the left side (receiving input from the right ear) dominant in most individuals. The sensory receptors of the vestibular system are in the ampullae of the semicircular canals (SCCs) and the otoliths (saccule and utricle). Similar to the cochlea, they contain hair cells, but with some important differences. The stereocilia connect to an overlying gelatinous structure called the cupula (similar to the tectorial membrane of the organ of Corti). Endolymph movement generated by head rotation pushes against the cupula, bending the hair cell stereocilia and thereby modulating excitation of the vestibular afferent fibres. The SCCs work in pairs so that when receptors in one ampulla are excited, those in the opposite ear are inhibited. Therefore, the afferent nervous output of each canal determines the magnitude of the rotation. The inner ear begins to develop at about 3–4 weeks. In the caudal part of the hindbrain, there is a thickening of surface ectoderm to form the otic placode. The ectoderm then invaginates to form a cup and then the otic vesicle, or otocyst. The otic vesicle separates from the overlying ectoderm and starts to elongate to become the cochlear duct, with an appendage which is the endolymphatic duct and sac (Fig. 31.4). The cochlear duct continues to grow ventrally from the 4th–8th week and begins to coil. Within the cochlear duct, the organ of Corti begins to develop from cells in the wall of the duct. Mesenchyme surrounding the cochlear duct condenses and differentiates into the cartilaginous otic capsule. This later ossifies to form the bony labyrinth around week 23. Ganglion cells of the VIIIth nerve migrate into the cochlear duct and form the spiral ganglion, sending projections to the ends of the hair cells and the organ of Corti. The inner ear is fully developed by 20–22 weeks. As the otic vesicle forms, the external and middle ear are also beginning their development just caudally. Development of the pinna of the external ear begins at about 5 weeks. It starts with the appearance of six mesenchymal swellings or hillocks, from the first and second branchial arches, surrounding the first branchial groove – from which develops the external auditory canal. Development begins in the upper part of the neck and as the mandible develops during the second month, the auricles move laterally and upwards level with the eyes. Migration of neural crest cells into the first and second branchial arches give rise to muscles and ligaments, and of importance here, the ossicles. The first arch neural crest cells give rise to the malleus and incus, and the second arch cartilage to the stapes. Genetically determined hearing loss can either exist as an isolated feature (non-syndromic) or as a syndrome in which it is associated with other abnormal clinical features. It is said that around 70% of genetically determined childhood-onset hearing loss is non-syndromic. Well over 100 genes are known to underlie non-syndromic hearing loss (NSHL) alone, and over 600 syndromes in which hearing loss is a feature have been described to date in the London Medical Databases (Dysmorphology and Neurogenetics) (see Further reading). Obviously, a paediatrician cannot be familiar with all of these syndromes, but some of the relatively common ones are listed in Table 31.1. It is helpful to be able to identify some of the more common syndromes involving hearing loss, to know how to approach investigation of a child with a suspected syndrome, and to be aware that some children who have apparent non-syndromic hearing loss may subsequently be determined as having a syndromic loss, as more clinical features become apparent with time. It is helpful to make these diagnoses in order to monitor for complications, and for the purposes of genetic counselling. For example, Pendred syndrome describes the association of congenital deafness and goitre, usually of teenage onset, which requires monitoring for subsequent hypothyroidism. A baby was born at 32 weeks to healthy unrelated parents following reduced movements in utero. She required continuous positive airway pressure (CPAP) for 24 hours, and had a 48-hour course of penicillin and gentamicin, which was discontinued following lack of bacterial growth on cultures and clinical improvement. She was discharged home at 3 weeks but failed newborn hearing screening. Bilateral profound hearing loss was confirmed at 2 months. At the second and third tier clinic, the paediatrician noted the combination of deafness, bright blue eyes, hair between the eyebrows (synophrys) and widely spaced inner canthi of the eyes (dystopia canthorum) suggesting a possible diagnosis of Waardenburg syndrome type 1. This was subsequently confirmed by a de novo mutation in PAX3. Non-syndromic hearing loss is very heterogeneous and may follow any mode of inheritance. Most genetic hearing loss is inherited in an autosomal recessive manner, accounting for about 80% of cases. The implication of this is that there may be no family history of deafness, but the risk of recurrence is high at 1 in 4. The remaining cases are either dominantly inherited (around 10–15%) or X-linked or mitochondrial (5% combined).
Hearing and balance
Anatomy of the ear
The external ear
The middle ear
The inner ear
The cochlea
The vestibular system
Structure and function
The process of hearing: converting sound into electrical energy
Central projections
Balance and the vestibular system
Embryology
The inner ear
The external and middle ear
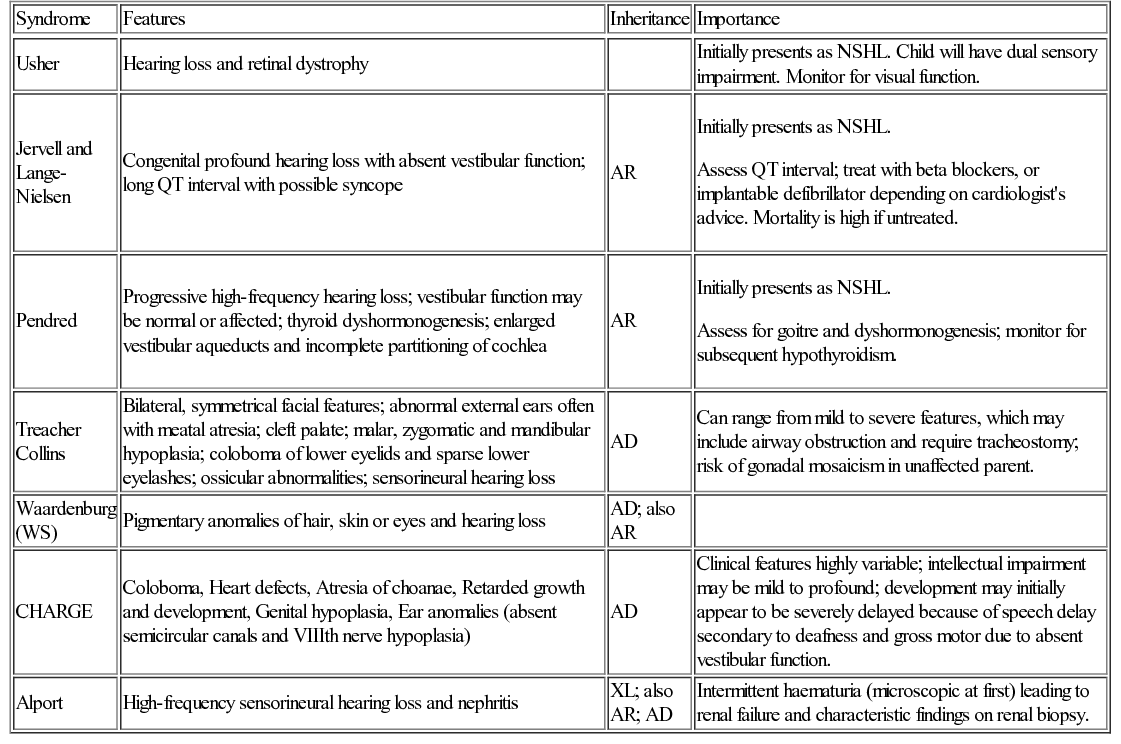
Genetic causes of hearing loss
 Case history
Case history
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hearing and balance
Chapter 31
Learning objectives
By the end of this chapter the reader should: