Mucopolysaccharidosis, Glycoproteinosis, and Mucolipidosis
James Edmond Wraith
MUCOPOLYSACCHARIDOSIS
The mucopolysaccharidoses (MPS) are a family of disorders that are caused by inherited defects in the catabolism of sulfated components of connective tissue known as glycosaminoglycans (GAGs). In affected patients, one or more of three specific polymers—dermatan sulfate (DS), heparan sulfate (HS), and keratan sulfate (KS)—accumulate within the cells, interfering with normal function, and are excreted in excess in the urine. 
The enzymes associated with GAG catabolism are all lysosomal hydrolases, and patients with an MPS disorder usually have less than 1% residual enzyme activity. Heterozygote detection based on enzyme activity alone is inaccurate and is now, fortunately, no longer necessary, as the genes encoding the enzymes involved in GAG catabolism have been identified and sequenced. Phenotypic variability (heterogeneity) is very much a feature of MPS disease, and within each specific enzyme deficiency there is a very wide spectrum of clinical effects. Although the disorders are most often known by their eponymous titles (eg, Hurler syndrome), this has led to an oversimplification in the classification of the subtypes, which should be kept in mind when interpreting the data in Table 160-1. A comprehensive review of the biochemistry and molecular biology of these disorders can be found in Neufeld and Muenzer, 2001.1
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
MPS disorders, like all lysosomal storage diseases, are progressive conditions. Affected infants are usually normal at birth, and the disease is suspected only as the phenotype evolves with time. Infants with an MPS-like phenotype present at birth are most likely to have mucolipidosis type II (I-cell disease) or GM1 gangliosidosis.
Table 160-1. The Mucopolysaccharidoses
The MPS disorders tend to present in one of three ways:
• As a dysmorphic syndrome (eg, MPS IH, MPS II, MPS VI)
• With learning difficulties, behavioral disturbance, and dementia (eg, MPS III)
• As a severe bone dysplasia (eg, MPS IV)
The diagnosis is based on clinical suspicion, supported by appropriate clinical and radiological examinations followed by urinary examination for GAG excretion and then by specific enzyme assay, usually on white blood cells. Urinary screening tests for MPS disorders may be inaccurate, and false-negative results, especially in MPS III and IV, are well recognized. Furthermore, patients with mucolipidosis or glycoproteinoses do not have excessive excretion of GAG. Therefore, a negative screening test should not dissuade the clinician from vigorously pursuing a diagnosis in a clinically suspicious case. Although algorithms aimed at helping the diagnostic process have been developed, their use in clinical practice is limited by the extreme heterogeneity in this group of disorders.
If urinary GAG analysis by electrophoresis is negative, one needs to consider other diagnostic possibilities. Urine oligosaccharide and sialic acid analysis should be undertaken to exclude oligosaccharidoses and other glycoproteinoses. White cell and plasma lysosomal enzyme studies should be performed to confirm abnormalities and to exclude galactosialidosis. Radiographs should be reviewed to confirm the presence of dysostosis multiplex (Fig. 160-1), and abnormal lysosomal storage should be confirmed by examining a skin biopsy under electron microscopy. If all these investigations are normal, it is important to remember that some nonlysosomal disturbances can mimic storage disease (eg, Coffin-Lowry syndrome, Williams syndrome, and geleophysic dysplasia).
All MPS disorders are multisystem diseases, and effective management depends on a multidisciplinary approach involving many different clinical specialties and access to expert support services. The pediatrician has a major role in orchestrating the various members of the therapeutic team. Anesthesia is particularly difficult in these patients, and surgery should be performed only in centers in which there is access to anesthetists who are used to dealing with difficult pediatric airways and who have access to pediatric intensive care. Many patients survive through adolescence and into adult life, and careful planning of the transition between pediatric and adult services is necessary.
There is a tendency to classify the individual MPS disorders into “mild” and “severe” subtypes, based either on survival or on the presence or absence of CNS disease. This is a gross oversimplification; it is preferable to consider the disorders on a clinical spectrum. Although many conditions are compatible with prolonged survival, for the majority of patients, these are not benign conditions.
MUCOPOLYSACCHARIDOSIS TYPE I
Patients with MPS IH (Hurler syndrome) are usually diagnosed toward the end of the first year of life, when the facial phenotype becomes obvious (Fig. 160-2). The younger a patient presents, the more likely he or she is to have a severe form of MPS I (Hurler syndrome).3 Many will have presented with inguinal and umbilical hernias and with recurrent respiratory infections before the diagnosis is established. Parents often notice the lower thoracicupper lumbar gibbus but are usually reassured that the abnormality is only postural and not of importance. A number of patients will have a large head circumference, and communicating hydrocephalus will develop in up to 40% of patients. The early clinical picture is dominated by the upper respiratory tract obstruction secondary to the midface hypoplasia, the large tongue, and the infiltration of the respiratory tract by accumulating GAG. Obstructive sleep apnea is usual, and all affected children require expert ear, nose, and throat (ENT) assessment; most require ENT surgery. Although growth for the first 12 to 18 months of life is normal, the effects of the skeletal dysplasia eventually lead to severe growth restriction. In addition, all patients have a dysplastic odontoid process and are at risk of sudden and severe spinal cord damage secondary to atlantoaxial subluxation (eFig. 160.2  ). Hepatosplenomegaly and progressive cardiac disease develops, and corneal deposition of GAG becomes clinically apparent as corneal clouding during the second year of life in most patients. Although learning difficulties are a feature of all patients with the severe forms of iduronidase deficiency, developmental progress can be surprisingly good over the first 2 or 3 years and often contrasts greatly with the affected child’s physical appearance. Deafness is usually present and must be diagnosed early and treated appropriately.
). Hepatosplenomegaly and progressive cardiac disease develops, and corneal deposition of GAG becomes clinically apparent as corneal clouding during the second year of life in most patients. Although learning difficulties are a feature of all patients with the severe forms of iduronidase deficiency, developmental progress can be surprisingly good over the first 2 or 3 years and often contrasts greatly with the affected child’s physical appearance. Deafness is usually present and must be diagnosed early and treated appropriately.
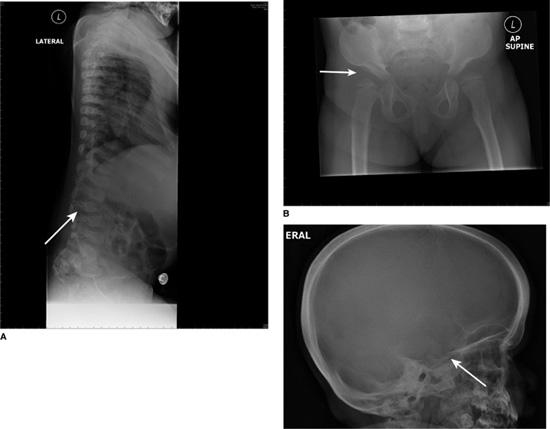
FIGURE 160-1. A: Dysostosis multiplex. Hypoplastic and hooked lumbar vertebral body (arrowed) at the site of thoracolumbar gibbus in a patient with MPS IH. B: Dysostosis multiplex. Shallow acetabulum and flattened femoral head (arrowed) in a patient with MPS IH. C: Dysostosis multiplex. Skull x-ray with thickened calvarium and J-shaped sella turcica (arrowed).
Although corneal clouding can be significant, severe visual loss is usually due to retinal or postretinal involvement. Sudden blindness can occur due to compression of the optic nerve within the optic sheath. Although glaucoma is said to be common, this is often over-diagnosed as increased corneal thickness gives rise to erroneous measurements of intraocular pressure. The ophthalmic and otolaryngological complications of the mucopolysaccharidoses have been reviewed recently.4,5
Prognosis depends upon the severity of cardiac involvement. This can range from a very severe cardiomyopathy, causing death in the early months of life, to progressive valve involvement (usually mitral and aortic) with relatively good left ventricular function and survival up to the end of the first decade and occasionally beyond. Coronary artery disease can be severe, and episodes of cardiac ischemia and infarction can occur. 
At the other end of the clinical spectrum from Hurler syndrome are those patients diagnosed in late childhood or early adult life, usually because of their orthopedic or oph-thalmologic problems. In MPS IS (Scheie syndrome), intellectual development is normal, and the disorder is compatible with a normal life span. Some patients require cardiac valve surgery, but the clinical picture tends to be dominated by bone and joint involvement. Carpal tunnel syndrome is almost universal in this type of MPS disorder. In some patients, corneal haze limits vision to such a degree that corneal transplantation is necessary. Most patients tolerate this procedure well, and the transplanted cornea remains clear. However, before a patient undergoes this surgery, a careful assessment of retinal function is necessary to ensure that the visual loss is not secondary to retinop-athy, which also occurs in MPS IS. The clinical presentation of this form of MPS I has recently been reviewed.6
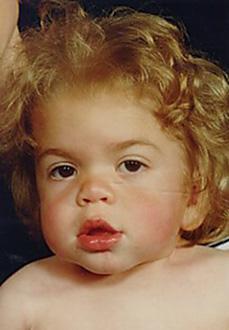
FIGURE 160-2. Facial features in MPS IH at diagnosis, age 12 months. Mid-face hypoplasia, “button” nose, thick lips.
Between these two extremes is a continuous clinical spectrum that is often labeled MPS IH/S (Hurler/Scheie syndrome; see Fig. 160-3). These patients may develop late-onset neurological deterioration, but most of the active clinical problems relate to the progressive joint stiffness and the degenerative bone disease that commonly occurs. Spondylolisthesis of L5/S1 is very common and requires surgical repair (eFig. 160.4  ). Progressive visual loss due to a combination of corneal clouding (eFig. 160.5
). Progressive visual loss due to a combination of corneal clouding (eFig. 160.5  ) and retinal disease is usual, and many patients will develop progressive cardiac disease. Frequent chest infections, limited chest expansion, and upper respiratory obstruction are common. In addition, many patients have a relative micrognathia (eFig. 160.6
) and retinal disease is usual, and many patients will develop progressive cardiac disease. Frequent chest infections, limited chest expansion, and upper respiratory obstruction are common. In addition, many patients have a relative micrognathia (eFig. 160.6  ) and are unable to open their mouths widely. Sleep apnea can be troublesome, and routine pulse oximetry overnight during sleep should be performed annually. Some patients require continuous positive airway pressure (CPAP) via a nasal mask. In patients who cannot tolerate the tight-fitting mask or the noise of the machine, tracheostomy remains the only alternative. In general, this is poorly tolerated in MPS disorders, and it is often associated with an increase in airway secretions that requires frequent suction.
) and are unable to open their mouths widely. Sleep apnea can be troublesome, and routine pulse oximetry overnight during sleep should be performed annually. Some patients require continuous positive airway pressure (CPAP) via a nasal mask. In patients who cannot tolerate the tight-fitting mask or the noise of the machine, tracheostomy remains the only alternative. In general, this is poorly tolerated in MPS disorders, and it is often associated with an increase in airway secretions that requires frequent suction. 
The gene coding for a-L-iduronidase is on chromosome 4p16.3 and consists of 14 exons. Many different mutations have been described, especially in the more severe forms of MPS I. Two nonsense mutations—p.W402X and p.Q70X—are relatively common in Europe, although their incidence varies from country to country. The molecular biology of MPS I has been reviewed recently.7
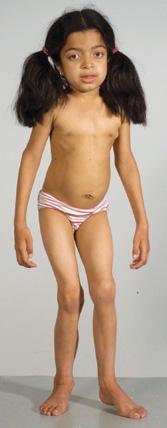
FIGURE 160-3. A patient with an intermediate form of MPS I (MPS IH/S, Hurler-Scheie disease). Note the joint stiffness and the relatively normal facial appearance.
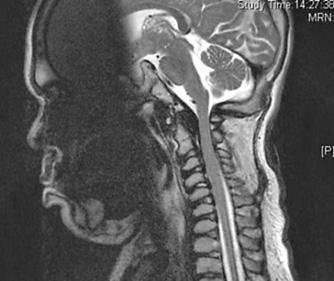
FIGURE 160-4. A lateral MRI scan of the craniocervical junction in a patient with MPS II. Notice the lack of CSF around the cervical cord (arrowed) due to thickening of ligaments and dura. The odontoid is also dysplastic.
MUCOPOLYSACCHARIDOSIS TYPE II
The clinical features in MPS II are even more heterogeneous than in MPS I. At the severe end of the clinical spectrum, the disorder is very similar to MPS IH but is generally milder, allowing for survival into the mid teens. At the other end of the spectrum, survival into adult life with reproduction is possible. There are two important differences from MPS I: First, the disorder is inherited as an X-linked recessive condition (it is the only X-linked MPS disorder; all the rest are recessives), and, second, corneal clouding does not occur to any significant degree in the vast majority of patients.
In severely affected patients, the diagnosis is usually established around the second birthday because of a combination of learning difficulties, middle-ear disease, a history of hernia repairs, and a coarse facial appearance. Many patients have troublesome diarrhea, and most develop joint stiffness and organomegaly. A nodular rash around the scapulae and on the extensor surfaces is considered pathognomonic of the disorder but is actually rare in childhood. The learning difficulties are different from those in MPS IH, as most patients with MPS II make more developmental progress than the typical MPS IH patient. The behavioral phenotype is also different, with challenging behavior, attention deficit disorder, and seizures being much more common in MPS II than in MPS IH. Cardiomyopathy (apart from asymmetric septal hypertrophy) is rare in MPS II, but progressive valve lesions can occur, although these rarely lead to symptoms.
In patients with less severe forms of MPS II, cervical cord compression due to hyperplasia of the dura and ligamentum flavum can lead to a progressive cervical myelopathy (Fig. 160-4). This usually presents with decreasing exercise tolerance that can be mistaken for a progression of the joint stiffness unless a careful neurological examination is performed. From the age of 10 years, these patients should have the craniocervical junction routinely evaluated by magnetic resonance imaging (MRI), and posterior decompression should be performed in all patients with cervical compromise. Fortunately, atlantoaxial instability is usually not a feature of MPS II, as the odontoid is usually well developed; therefore, spinal fusion in addition to the decompression is usually not required.
Despite often gross abnormality on cerebral imaging, intellectual development is normal in this form of MPS II, but the severe somatic features present in some intellectually normal men lead to considerable psychosocial problems, which require sensitive and careful handling.
Most adults with MPS II develop upper respiratory obstruction and sleep apnea. Many benefit from the use of nasal CPAP devices, although the nasal masks often have to be shaped individually because of the abnormal facial anatomy.
Although MPS II is an X-linked disorder, an occasional affected female patient has been described. This can occur as a result of chromosomal translocation or nonrandom X-inactivation. Most affected girls have an intermediate phenotype with preserved cognitive function (eFig. 160.7  ).
).
The MPS II gene is large and is located at Xq27-28. In affected boys, a whole range of molecular pathologies have been described, including insertions, deletions, point mutations, and splice-junction mutations. There are no common mutations, and it is often difficult to predict severity from the molecular lesion. In some patients, no abnormalities are detected despite sequencing of the whole coding region, and it is assumed that other regulatory elements must be involved in the disease. The molecular biology of MPS II has been recently reviewed.9
MUCOPOLYSACCHARIDOSIS TYPE III
This MPS disorder is a clinically similar but biochemically heterogeneous group of four recognized conditions all associated with an inability to catabolize heparan sulfate. MPS IIIA is the most common MPS disorder in the UK (and many other countries) and accounts for 80% of all MPS III patients in this population. The remaining patients are mainly MPS IIIB; types IIIC and IIID are rare. 
The hallmark of MPS III is severe central nervous system (CNS) involvement in the presence of a mild somatic phenotype. Because of this combination, the diagnosis is usually established much later in life (4-5 years),12 when compared to other MPS disorders, and the condition is less heterogeneous than MPS I or II. In a typically affected patient, a triphasic illness can be recognized. The first phase, often before diagnosis, consists of only developmental delay. There is often a history of recurrent upper respiratory infections, and most patients have troublesome diarrhea. Sleep disturbance can present early in life, and in its extreme form produces a reversal of the normal sleep/wake cycle.
Gradually the characteristic behavioral phenotype evolves as the second phase of the illness starts, usually in late infancy. This comprises severe challenging behavior with extreme hyperactivity and often aggression. In addition, there is a complete disregard for danger, and the children are a risk to themselves and others and need constant attention. Temper tantrums are frequent, and normal family life for many families becomes impossible. It is during this phase that the diagnosis is established in the majority of patients. As the disease advances, developmental milestones are lost and increasing spasticity leads to progressive loss of motor skills. Precocious pubertal development is a well-recognized association.
The third and final stage of the illness usually begins in the early teenage years and is characterized by further loss of skills leading to swallowing dysfunction; eventually the disorder culminates in a vegetative existence in the mid-to-late teens. Death usually occurs around the second decade, although patients with MPS IIIC may have a more attenuated course and can survive in to the third or fourth decade.
Seizures are common in the later stages in some patients and can be difficult to control, while other patients develop a severe movement disorder resistant to treatment. Mood disturbance with prolonged crying can be extremely distressing for the parents of affected children.
Somatic features are usually mild (Fig. 160-5), except in some patients from the Asian subcontinent who can develop severe cardiac involvement (usually mitral valve disease but occasionally a dilated cardiomyopathy).
The disorder is probably underdiagnosed at the less severe end of the clinical spectrum, as these patients may have only mild learning difficulties until the age of 20 to 30 years.
Mutation analysis has been performed extensively in MPS IIIA and IIIB. The MPS IIIA gene is located on chromosome 17q25.3 and consists of eight exons. Considerable genetic heterogeneity has been demonstrated and a wide range of mutations described; p.R245H and p.R74C have a combined frequency of over 50% in some European populations. The MPS IIIB gene is situated on chromosome 17q21.1 and consists of six exons. An even greater degree of genetic heterogeneity is seen in this disorder, and there are no common mutations.
The MPS IIIC gene has recently been cloned,13 and the MPS IIID gene is of academic interest only because so few patients with MPS IIID have been identified. 
MUCOPOLYSACCHARIDOSIS TYPE IV
Patients with MPS IV have a severe skeletal dys-plasia and, unlike the other MPS conditions, are not dysmorphic. In addition, CNS involvement does not occur, and the clinical course is dominated by the severe bone disease and resulting extreme short stature. The diagnosis can be established in the newborn period but more commonly is made toward the end of the first year of life, when the sternal protrusion, which occurs as a result of the short trunk, becomes obvious. The radiological abnormalities are different from the classic dysostosis multiplex seen in MPS I, II, VI, and VII and are characterized by vertebral platyspondyly and other features of a generalized spondyloepiphyseal dysplasia (see Fig. 160-6).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


