Elisabeth Jameson • Understand the biochemistry of metabolism, including urea cycle, Krebs cycle and fatty acid cycle • Know the genetic and environmental factors in the aetiology of metabolic disorders • Be aware of the metabolic disorders identified on neonatal screening • Understand the investigations that are used to diagnose metabolic disorders • Understand the principles of dietary and pharmacological treatment of metabolic diseases Intermediary metabolism is the term given to the biochemical reactions that degrade, synthesize or interconvert molecules within the cells. There are numerous metabolic pathways, which serve the following aims: These pathways require enzymes, which, if absent or deficient, can give rise to an inborn error of metabolism (IEM). This chapter describes the key metabolic pathways and links them with their associated diseases. The key terms are outlined in Table 29.1. Table 29.1 Acid–base definitions Consists of a weak acid in the presence of its base. A buffer serves to minimize changes in H+ concentration in response to the addition of an acid or base. Examples of buffers in: Plasma – bicarbonate, proteins, inorganic phosphate (Pi) Erythrocytes – haemoglobin, bicarbonate, Pi Kidneys – bicarbonate, Pi, ammonium Acid–base balance is essential for correct cellular functioning. Blood gas measurement can identify the primary disturbance (Table 29.2). In general: In the case of metabolic acidosis, calculation of the anion gap will determine if there is the presence of an unmeasured anion such as an organic acid, e.g. methylmalonic or propionic acid (Table 29.3 and Fig. 29.1). Acidosis with a normal anion gap is often associated with hyperchloraemia because the loss of base is buffered by an increase and/or retention of chloride. Metabolic acidosis is a common finding. In the majority of cases, it reflects severe illness rather than an inborn error of metabolism (IEM). The latter should be considered if the acidosis is out of keeping with the clinical picture, is persistent despite standard management and there is no identifiable acid present, e.g. lactate or ketones. Metabolic acidosis is typically non-specific in presentation. Signs may include a reduced conscious level, vomiting or those associated with the underlying aetiology, e.g. non-blanching rash in the case of meningococcal sepsis. Many patients will display an increased respiratory rate, Kussmaul respiration, reflecting the compensatory hyperventilation that occurs to promote removal of carbon dioxide. The blood gas is key to identifying the primary disturbance in acid–base balance. In addition to calculating the anion gap, ketones and lactate should be measured as potential causes of acidosis. When investigating for an IEM, urine organic acids and plasma amino acids and acylcarnitines are required. It is important to measure an ammonia level as this can be elevated in an organic acidaemia due to the metabolites inhibiting the urea cycle. The underlying aetiology, when known, should be treated. If acidosis is severe, normalization of acid–base balance can be achieved with administration of sodium bicarbonate. Normal plasma lactate is <2 mmol/L. A raised level has a wide differential (Table 29.4). In terms of IEM, mitochondrial disorders are classically associated with a raised lactate, with levels often fluctuating. When considering the possibility of mitochondrial disease, measuring cerebral spinal fluid for a raised level can be helpful. However, a normal lactate does not exclude a mitochondrial disorder. Key points – organic acid disorders • Methylmalonic and propionic acidaemia are the most common organic acidaemias • Can cause pancytopenia because of effects on the bone marrow at times of decompensation • pH can be maintained with hyperventilation • A lactate of 8 mmol/L would not by itself generate such a large anion gap • To calculate a half correction (using this case as an example):
Metabolic medicine
Introduction
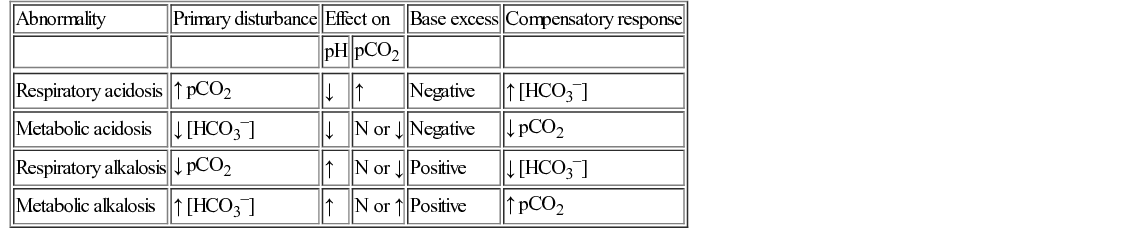
Acid–base disturbance
Definitions
Terminology
Definition
Acid
A proton or hydrogen ion donor. It can dissociate to yield H+ and the corresponding base.
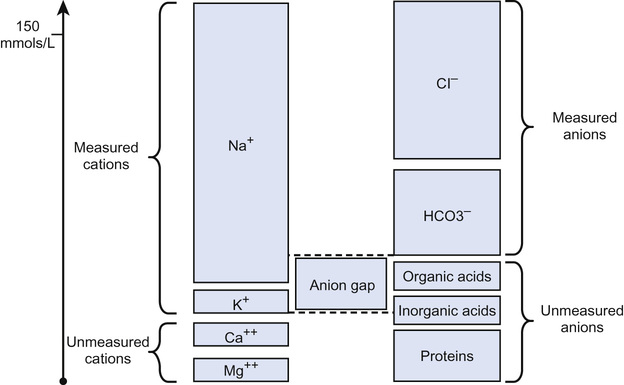
Anion gap (see Fig. 29.1)
[Na+ + K+] − [Cl− + HCO3−]. Normal = 10 − 16 mmol/L. Reflects concentration of those anions not routinely measured, e.g. organic acids (see Question 29.1).
Base
A proton or hydrogen ion acceptor. Can accept H+ to form corresponding undissociated acid.
Base excess
Measures the change in the concentration of a buffer base from the normal value. Normal range = +/− 2 mmol/L.
Buffer
pH
The logarithm to the base 10 of the reciprocal of the hydrogen ion concentration. pH = −log [H+]
pKa
The pH of a buffer at which half the acid molecules are undissociated and half are associated.
Biochemistry
Clinical
Presentation
Diagnosis
Management
Lactic acidosis
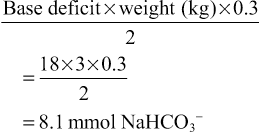


The urea cycle and hyperammonaemia
Biochemistry
Ammonia
Ammonia is a highly neurotoxic chemical detoxified by the urea cycle (Fig. 29.2), which principally occurs in the liver. Ammonia is formed from:
• Nitrogen produced from amino acid metabolism
• Glutamate by the action of glutamate dehydrogenase
• Glutamine by the action of glutaminase.
• Alanine and glutamine produced by muscle turnover
• Urease-positive gut bacteria
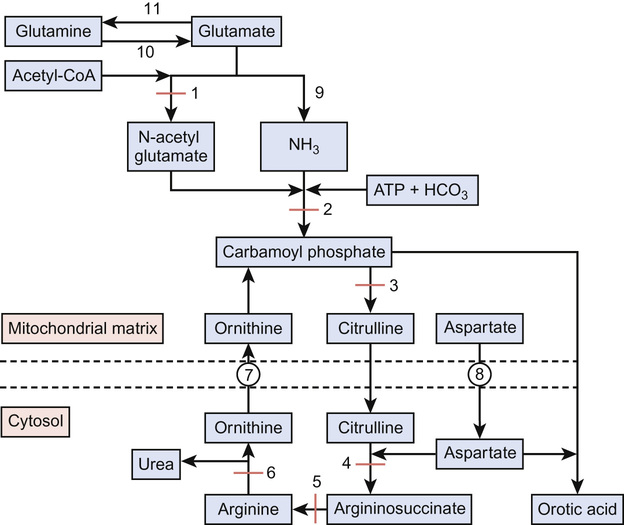
The urea cycle
The urea cycle (see Fig. 29.2) consists of six enzymes, with each full cycle disposing of two nitrogen atoms: one from ammonia and one from aspartate. The cycle progresses as:
• Carbamoyl phosphate condenses with ornithine to form citrulline
• Argininosuccinate lyase cleaves argininosuccinate to arginine
Ammonia is also buffered by the conversion of glutamate to glutamine via the action of glutamine synthetase. At times of hyperammonaemia, the glutamine concentration increases and thus can be used as an indicator of insufficient urea synthesis and is indicative of longer term metabolic control.
Clinical
Hyperammonaemia (normal plasma ammonia levels are <100 µmol/L in neonates and <50 µmol/L thereafter) has a wide differential (Table 29.5). Urgent measurement of ammonia should therefore take place in any baby, child or adult presenting with unexplained encephalopathy or illness. The urea cycle disorders (UCD) arise due to deficiency of one of the six main urea cycle enzymes.
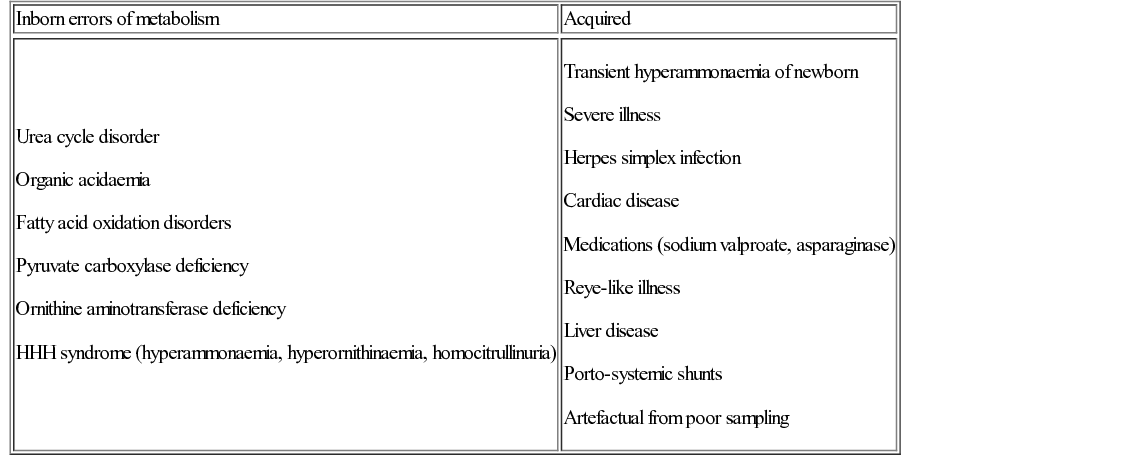
Presentation
The classic presentation is the term baby who becomes increasingly sleepy and encephalopathic on day 3–5 of life with poor feeding and vomiting (see Question 29.2). Ammonia levels can rise rapidly. Urgent investigation is required to clarify the diagnosis and guide management.
The urea cycle disorders are inherited in an autosomal recessive manner, except for ornithine transcarbamylase (OTC) deficiency, which is X-linked (see Genetics of metabolic disorders, below). Male infants are severely affected and many do not survive the neonatal period. Female carriers have a varied phenotype; the majority remain asymptomatic but approximately 15% will require treatment.
Diagnosis
Diagnosis of urea cycle disorders (Table 29.6) is based upon plasma amino acid analysis and the presence or absence of urine orotic acid, which is produced when carbamoyl phosphate passes into the pyrimidine pathway. The absence of orotic acid in a urea cycle disorder implies N-acetylglutamate synthetase (NAGS) or carbamoyl phosphate synthetase (CPS) deficiency. Orotic acid is classically very elevated in OTC because of the accumulation of intracellular carbamoyl phosphate. The remaining defects are associated with a much smaller or negligible amount of orotic aciduria.
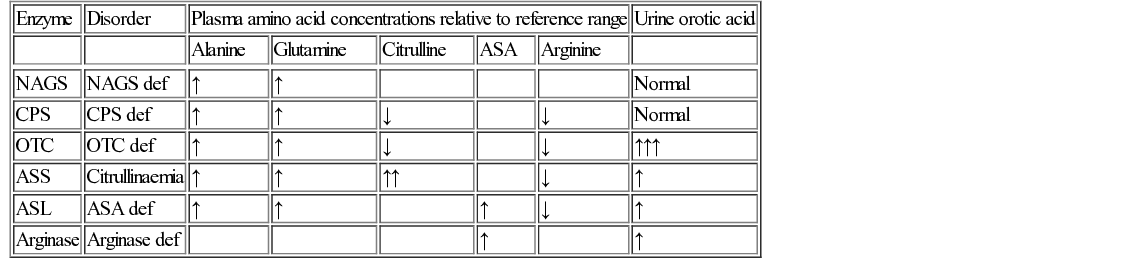
Management
This can be thought of in terms of acute and long term.
Acute:
• Stop feeds and commence 10% dextrose to reduce nitrogen load on the cycle
• Commence intravenous ammonia scavenging medications (see Principles of pharmacological treatment, below)
• Commence intravenous arginine to replenish the urea cycle
• Transfer to specialist centre in preparation for haemofiltration
Chronic:
• Low protein diet to reduce nitrogen load on the cycle
• Ammonia scavenging medications to aid excretion of excess nitrogen
• Arginine (except in arginase deficiency) to replace arginine not produced by the urea cycle



