Metabolic Consequences of Sleep Disordered Breathing
Introduction
Epidemiological studies over the last several decades have conclusively demonstrated that both the prevalence and severity of overweight and obesity in children and adolescents have clearly been increasing worldwide, even if some deceleration in such trends has emerged more recently, possibly as the result of public health campaigns aiming to curb the alarming rates of obesity in childhood.1–4 Nonetheless, reported prevalences of overweight hovering around 10.4% among toddlers, 15.3% among school-aged children and 15.5% among adolescents are now frequently exceeded in many of the underserved communities across the USA and the world.5–8 A major consequence of this epidemic of obesity has been the concomitant increase in the prevalence of obesity-associated morbidities, such that previously infrequent conditions such as the metabolic syndrome, cardiovascular disease, non-alcoholic liver steatosis, depression, and decreased quality of life have all begun to emerge, even among the youngest of the children.9–12
Sleep Curtailment, Irregular Sleep, and Obesity
The increasingly demanding life pace of technology-driven societies has radically changed the way we sleep, as well as the way our children sleep. Over a period of 100 years or so, the overall duration of sleep in children has steadily declined.13–15 Although the reasons underlying these trends in sleep curtailment are multifactorial,16–18 reductions in sleep duration have also been accompanied by increased irregularity in sleep schedules. There is now compelling evidence linking the progressive decrements in sleep duration and sleep regularity to the reciprocal increases observed in the prevalence of childhood obesity.19–28 However, the associations between obesity and sleep have not been consistently reported by all studies,29 raising the possibility that the current definitions of short sleep and insufficient sleep are quite arbitrary, and therefore operate as a confounder rather than a true causal pathway.30–32 Furthermore, additional confounding factors that play a role in the propensity for obesity may begin in early life,33 such that extrication of the role of sleep may not be trivial. In a cross-sectional and longitudinal study, Chaput and collaborators reported that only those subjects who exhibited the combination of short sleep duration, high disinhibition eating behavior, and low dietary calcium intake had significantly higher BMI.34 Based on the aforementioned considerations, it is clear that intervention trials aiming to establish whether restoration of sleep quality and quantity will ameliorate metabolic and ponderal indices in children are needed.35 It is also noteworthy that sleep-associated changes in BMI appear to be primarily affecting those children whose BMI is already elevated.36
Some of the putative biological pathways linking sleep duration and regularity to obesity clearly involve alterations in some of the neuropeptides that regulate appetite both peripherally and centrally. In addition, the contribution of perturbations in circadian timing cannot be overlooked.37 For example, increased levels of ghrelin, reduced levels of leptin, and reduced central biological activity of orexin have all been identified in experimental sleep restriction paradigms, and will lead to increased food intake.38,39 Furthermore, disruptions of the circadian clock have been implicated in perturbations within a complex network of metabolic pathways that not only affect regulatory CNS regions such as in the hypothalamus, but also alter metabolic processes in peripheral tissues.40,41
Unfortunately, in-depth exploration of these pathways in the context of childhood is almost completely lacking; furthermore, we should keep in mind that both sleep duration and body weight trajectory are determined by a multitude of factors, such as sociodemographic, socioeconomic, familial (e.g., family structure, overweight parent) and individual (e.g., health behavior, physical activity, health status, stress levels) factors (Figure 31-1).42–44 Regardless, we need to be cognizant that obese children are 1.5- to 2-fold (odds ratios ranging from 1.15 to 11) more likely of being short sleepers,45,46 and that any interventions seeking to modify sleep patterns in children are less likely to succeed if implemented later in childhood, since both sleep regularity and sleep duration are consistently preserved across long periods of time even in children.47 Thus, sleep interventions need to be implemented early in life, and therefore identification of children at risk during infancy and early childhood and prospective interventions to prolong and regularize sleep are required to delineate the role of sleep in the context of BMI regulation and metabolic homeostasis.
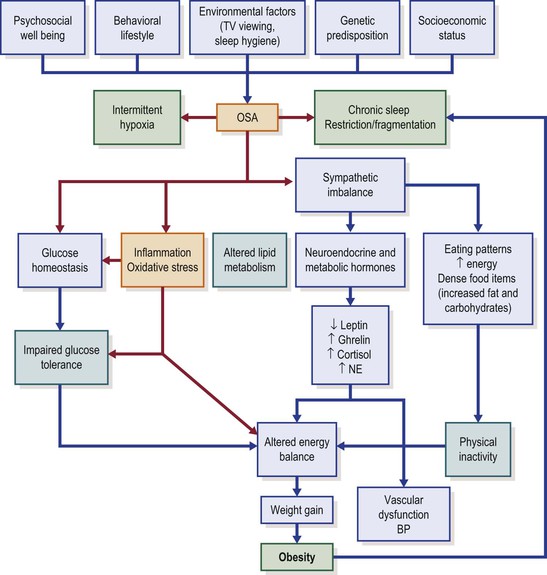
Obesity as a Risk Factor of Obstructive Sleep Apnea
Since the early descriptions of obstructive sleep apnea (OSA) in the pediatric age range, a clear change has occurred, with a majority of the patients being evaluated in most pediatric sleep centers fulfilling the criteria for either overweight or obese.48 This is not surprising, considering that the risk of OSA in obese children is markedly increased.49 In a case-control study design, Redline and colleagues examined risk factors for sleep-disordered breathing in children aged 2–18 years, and found that the risk among obese children was increased 4–5-fold.50 In fact, for every increment in BMI by 1 kg/m2 beyond the mean BMI for age and gender, the risk of OSAS increased by 12%. Similar trends demonstrating increased risk of OSA among obese and overweight children have been consistently reported in many countries.51–60
Upper airway narrowing in obese children could result from fatty infiltration of upper airway structures and tongue, while subcutaneous fat deposits in the anterior neck region and other cervical structures will undoubtedly contribute and exert collapsing forces promoting increased pharyngeal collapsibility.61,62 Interestingly, Arens and collaborators have recently shown that obese children exhibit enlarged lymphadenoid tissues in the upper airway, the latter potentially reflecting the result of systemic low-grade inflammatory processes that accompany the obese state.63,64 Obesity may also affect lung volumes through mass loading of the respiratory system,65 and increased adipose tissue deposition in the abdominal and thoracic walls and around the viscera will increase the overall respiratory load, and reduce intrathoracic volumes and diaphragmatic excursion, particularly when in the supine position. Thus, obesity will lead to decreased lung volumes and oxygen reserve, and will augment the work of breathing during sleep.
Obesity is also associated with peripheral and central leptin tissue resistance, resulting in reduced biological effectiveness despite elevation of circulating leptin levels.66–68 The unique properties of leptin as a potent respiratory stimulant with both central and peripheral chemoreceptor modulatory properties are therefore markedly dampened by the emergence of leptin resistance, and may precipitate the gradual onset of attenuated respiratory reflexes, particularly during sleep.69,70 Obesity can be accompanied by fragmented sleep that will progressively increase the arousal threshold, thereby potentially aggravating the duration of upper airway obstruction events.71
OSA May Promote Obesity and Its Consequences
Although there is currently only incipient evidence, it is plausible to assume that the presence of OSA could promote or aggravate obesity or its consequences (see Figure 31-1). This concept has developed in recent years whereby the presence of OSA would promote weight gain and obesity, via sleep fragmentation and associated daytime sleepiness.72–75 The presence of sleepiness is likely to reduce overall daily physical activity,76 particularly in those children at risk for obesity. In addition, the degree of daytime sleepiness resulting from OSA is amplified in obese children.77
Similar to obesity, OSA should nowadays be viewed as a low-grade systemic inflammatory disease, and the coexistence of obesity and OSA further exacerbates the magnitude of the inflammatory responses.78–83 Thus, OSA is viewed as a major risk factor for development of the metabolic syndrome in children (see also Chapter 18).
OSA Potentiates Alterations in Serum Lipids in Children
In a large series of studies in adult patients, OSA has been identified as a risk factor for dyslipidemia and insulin resistance.84–86 However, since most adult OSA patients are obese, it is nearly impossible to determine the exact contribution of OSA on lipid metabolism.
The evidence in children linking OSA to lipid alterations is relatively scarce. In a study of 135 children (many of them being obese), no associations emerged between polysomnographic indices of OSA severity and fasting serum lipids, even though strong association was apparent between BMI and lipid levels.87 Verhulst et al.88 showed significant correlations between the degree of oxyhemoglobin desaturation (but not the respiratory disturbance index) and serum lipid and cholesterol levels among 104 relatively older obese children (42% post pubertal), even when controlling for gender, puberty and body mass index. In a retrospective study of Greek children being evaluated for suspected OSA, Alexopoulos and colleagues89 reported that the risk of low HDL cholesterol was 3-fold higher in non-obese children with moderate to severe OSA compared to those children with mild OSA or primary snoring. In a prospective cohort study of adolescents recruited from the general community, Redline et al.90 reported a 6-fold increase in the risk for developing the metabolic syndrome in those individuals with OSA. Furthermore, OSA in these adolescent subjects was closely associated with serum LDL concentrations. In a more recent study, Zong and collaborators further confirmed the existence of putative inverse associations between HDL cholesterol and AHI in a small cohort of 44 children in China.91
In a prospective study of 62 consecutive pre-pubertal obese and non-obese children with OSA, surgical adenotonsillectomy resulted in improvements of OSA in all children, but normalization of the respiratory disturbances during sleep occurred only in a fraction of these children. In non-obese children, surgical removal of adenoids and tonsils led to significant improvements in LDL, HDL, and LDL/HDH levels, and overall similar findings occurred in obese children with OSA after treatment, in the absence of any significant changes in BMI. These findings suggest that the improvements in serum lipids can be ascribed to OSA rather than obesity. Apolipoprotein B serum levels were improved in both non-obese and obese groups following surgery, and all lipids were correlated with measures of OSA severity.92 Taken together, these findings strongly support that disrupted sleep and episodic hypoxia in the context of OSAS impose substantial changes in lipid regulatory mechanisms in children.
Since not every child with OSA will manifest alterations in serum lipids, it is possible that genetic factors may contribute. To examine this issue, Bhushan and colleagues93 assessed gene variants in the fatty-acid-binding protein 4 (FABP4) gene, a gene that plays a major role in inflammation and metabolism, particularly in the regulation of the intracellular processing of lipids in critical tissues such as adipose tissues and macrophages. Children with OSA had elevated concentrations of FABP4 in plasma, and the presence of the rs1054135 polymorphism in the FABP4 gene was associated with elevated levels of FABP4 and serum lipids, even after adjustment of BMI, suggesting that genetic predisposition may contribute to impaired lipid homeostasis in children with OSA.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


