Apnea of Prematurity
Pathophysiology
The Role of Upper Airway Obstruction
Traditionally, apnea is divided into central, obstructive and mixed. Many apparently ‘central’ apneas, however, involve a loss of airway tone that results in intermittent airway obstruction,1 while active glottic closure, similar to that preventing outflow of lung water during in utero apnea,2 has also been observed in AOP, potentially preserving lung volume during apnea also ex utero. Airway obstruction may also prolong initially short respiratory pauses.3 Thus, the narrow upper airways of preterm infants might be actively maintained open via a respiratory center input, and it simply depends on which component of this input is activated first (diaphragm or upper airway) whether an apnea will appear as central or obstructive.4
Relationship between Apnea, Bradycardia and Desaturation
Apnea, bradycardia and desaturation during AOP are closely temporally related.5 According to the author’s data on 80 preterm infants, 83% of bradycardias (heart rate less than two-thirds of baseline) were accompanied by an apnea (≥4 s), 86% by a fall in pulse oximeter saturation (SpO2) to ≤80%, and 79% by both apnea and desaturation.6 The interval between apnea and bradycardia was extremely short (median, 4.8 s), as was that between bradycardia and desaturation (median, 4.2 s). This was predominantly because the interval between the onset of apnea and that of desaturation, corrected for the time it takes for the blood to travel from the lung to the pulse oximeter sensor site, was only 0.8 s (Figure 24-1).6
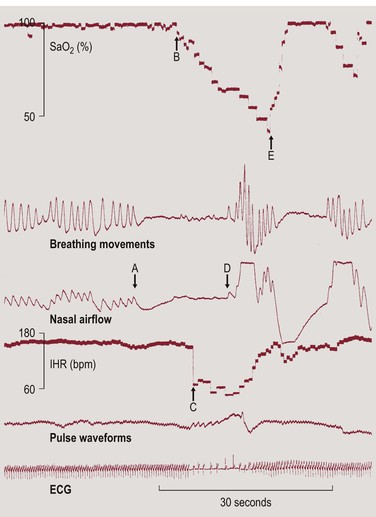
Figure 24-1 Example for the close temporal relationship between apnea, bradycardia and desaturation. The delay caused by the time it takes for the blood to travel from the lung to the pulse oximeter sensor attached to the foot can be estimated from the delay between the first breath following an apnea and the onset of the recovery in SpO2 (D to E). This must be subtracted from the interval between the onset of apnea and that of desaturation (A to B) and from the interval between the onset of bradycardia and that of desaturation (B to C). Reproduced from Poets CF. Pathophysiology of Apnea of Prematurity: Implications from Observational Studies. In: Mathew OP. Respiratory control and its disorders in the newborn. Marcel Dekker, Inc, New York 2003,295–316, with permission.
These observations support the concept that hypoxemia causes bradycardia, e.g., via stimulation of peripheral chemoreceptors,7 the occurrence of which is facilitated by the absence of the pulmonary inflation reflex during apnea. The latter would also explain why, despite a similar severity of the accompanying hypoxemia, bradycardia is more common with central than with mixed or obstructive apnea.8
Changes in Lung Volume, Apnea and Desaturation
Why is the interval between apnea and desaturation onset so short? This may be related to a reduced functional residual capacity (FRC). In preterm infants, relaxation volume is only 10–15% of total lung capacity and thus very close to residual volume, predisposing them to the development of peripheral airway closure.9 To compensate for this disadvantage, these infants actively maintain their end-expiratory lung volume above relaxation volume, which is one reason for their high respiratory rate.10 In fact, lung volume is 20% lower after an apnea than after a sigh, i.e., apnea results in a loss of FRC, which is restored by a sigh. This suggests that one of the main functions of sighs in preterm infants is to reverse falls in lung volume caused by apneas.11,12
During periodic apnea, SpO2 falls twice as fast as during isolated apneas.5 Although a reduced mixed venous SO2 following a prior fall in SpO2 may also contribute to this, another reason for this is a progressive fall in lung volume during the repeated apneas, resulting in peripheral airway closure. The complex interrelations of the factors influencing the speed of the fall in SpO2 during AOP have recently been modeled mathematically.13
A loss in lung volume may also be decisive for the development of hypoxemia following abdominal muscle contractions in ELGANs, being involved in 80% of their desaturations to <75% SpO2 in one study, and being associated with an average decrease in resting lung volume by 69% of tidal volume.14
A potential consequence of a reduction in lung volume is a (further) inhibition of respiration via activation of the Hering–Breuer deflation reflex. In term infants, this vagally mediated reflex terminates expiration while initiating inspiration. In preterm infants, however, induction of this reflex via chest compression resulted in a shortening of inspiratory time and a tendency to have short apneas (2–5 s).15 The same may occur if lung volume falls spontaneously, e.g., during apnea.
Conversely, an increase in lung volume was recently found to stabilize breathing by reducing loop gain, i.e., the sensitivity of the negative feedback loop of the chemoreflex control of the respiratory system, while a reduced lung volume will increase the instability of the respiratory control system, as evident during periodic breathing.16
These considerations provide a theoretical basis for the effectiveness of strategies that increase lung volume in reducing the frequency and/or severity of AOP in preterm infants.17
The Role of Feeding and Gastro-Esophageal Reflux
Symptoms of AOP often increase in relation to feeding. We studied the effect of bottle feeding, as compared to slow (1 h) and bolus (10 min) gavage feeding, on AOP and found three times more desaturations to ≤80% with bottle than with bolus gavage feeding, but no further reduction with slow gavage feeding. There were always significantly more desaturations in the hour feeds were given than during the following 2 hours, but the effect of feeding method persisted throughout these 3-h feeding intervals. Interestingly, there was no significant effect of feeding technique on the frequency of apnea or bradycardia (Figure 24-2).18
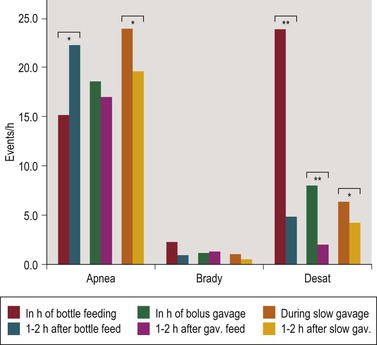
Figure 24-2 Apnea, bradycardia (Brady) and desaturation (Desat) rates during the hour feedings were given compared to the following 2 hours in 30 preterm infants studied at 34 wk (SD 1.4) gestational age. * p < 0.05, ** p < 0.01. Reproduced from Poets C. Apnea of Prematurity. In: Kheirandish-Gozal L, Gozal, D., editor. Sleep-Disordered Breathing in Children: A Comprehensive Clinical Guide to Evaluation and Treatment. New York: Humana press, Springer; 2012. p. 263–78, with permission.
With the new multiple intraluminal impedance (MII) technique, GER can now be detected, independent of acidity, via changes in impedance caused by a liquid bolus inside the esophagus. We recorded MII, together with cardiorespiratory (CR) signals, in 19 infants with AOP.19 MII signals were analyzed, independently of CR signals, for reflux episodes. CR signals were analyzed for apneas (≥4 s), desaturations to ≤80%, and bradycardias to ≤100 bpm. A temporal relationship between GER and a CR event was considered present if both commenced within 20 s of each other. We found a high rate of both apnea and GER, but the apnea frequency within 20 s of a reflux episode was not significantly different from that during reflux-free epochs (mean, 0.19/min. vs. 0.25/min); the same was true for desaturations and bradycardias. Also, GER occurred similarly often before as after an apnea. Only in the few apneas (3.5%) that were associated with GER reaching the pharyngeal level, significantly more (45 vs. 26) occurred after rather than before GER. Thus, while both CR events and GER were common, they did not, with few exceptions, appear to be temporally related.19 Similar results have since been reported by others.20,21 Thus, the widespread practice22 of giving antireflux medications to infants with AOP is futile.
Chest Wall Distortion, Anatomic Dead Space and Diaphragmatic Fatigue
Chest wall distortion, clinically apparent as paradoxical breathing, is common in infants and is especially visible in preterm infants. It has been suggested that this distortion increases the volume displacement of the diaphragm during inspiration.23,24 In longitudinal studies, Heldt showed that the minute volume displacement of the diaphragm was almost twice as large as pulmonary ventilation at 29–30 weeks’ gestational age (GA) and fell to approximately 90% of pulmonary ventilation at 36 weeks’ GA. Concomitantly, diaphragmatic work was almost halved.24 The author speculated that this additional workload may represent not only a significant calorie expenditure in these infants, but contributes to the development of diaphragmatic fatigue and apnea.24 Further contributing is that, because of their relatively large head size, anatomic dead space is approximately 45% of tidal volume in neonates, but only 25% in adults.25,26
Circumstantial evidence that muscle fatigue may indeed be involved in neonatal apnea stems from the time course of apnea in term and preterm infants. AOP becomes more problematic towards the end of the first or during the second week of life,27 while chemoreceptor resetting, which otherwise might also explain this phenomenon, is essentially complete within approximately 24–48 h after birth.27 We confirmed this observation for desaturation rates in both term and preterm infants.28,29 A mechanism through which labored breathing may produce apnea in preterm infants is the intercostal–phrenic inhibitory reflex. This may be elicited both by rib cage distortion30 and respiratory loading,31 and is known to inhibit respiratory effort in infants.
Hypoxic Ventilatory Depression
Fetal breathing is diminished if oxygen supply via the placenta is reduced.32 For the fetus, respiratory movements are a waste of energy that cannot be afforded if oxygen supply via the placenta is reduced. This behavior, however, although counterproductive ex utero, continues after preterm birth,33,34 switching to a mature hypoxic ventilatory response only at approximately 35 weeks’ PCA. This correlates well with the natural course of AOP.35 While the classic hypoxic response is biphasic, consisting of an initial increase followed by a decrease in ventilation,33,35 ELGANs show an immediate reduction in minute ventilation during hypoxia, which is mainly due to a fall in respiratory rate.36
The clinical relevance of this maturational phenomenon was recently demonstrated in a substudy of SUPPORT, a large multicenter study comparing the effects of a high (91–95% SpO2) versus a low (85–89%) target range for SpO2 in ELGANs.37 By recording desaturation rates until 36 wk PCA in infants randomized in their center, the authors found significantly (2–3 times) more desaturations to ≤80% for ≥10 s in the first 11 days of life and again between 8 weeks of age and reaching 36 wk PCA in those infants who had been randomized to the lower SpO2 target group.38 This observation is best explained by the effects seemingly small decreases in baseline SpO2 have on hypoxic ventilatory depression.39
As mentioned above, an important question is which molecular mechanisms are responsible for the hypoxic ventilatory depression. One candidate is the creatine–phosphocreatine (PCr) system. In the absence of oxidative phosphorylization, provision of phosphate for generation of adenosine-triphosphate (ATP) relies predominantly on the PCr pool, before anaerobic glycolysis, with increased production of lactate and H+, is activated.40 This is particularly relevant to tissues with a high energy metabolism such as the central nervous system. A fall in intracellular ATP is an important trigger for hypoxia-induced neuronal damage, and maintenance of ATP levels is therefore of fundamental importance for neuronal protection from hypoxic insult.41 The neonatal brain is relatively deficient in creatine, and it is tempting to speculate that the much earlier onset of the hypoxic ventilatory depression in this age group is related to a decreased availability of PCr in the neonatal brainstem.
Brainstem slices from pups of creatine-fed mice (2 g/kg/d) did indeed show higher phosphocreatine contents and significantly less hypoxic ventilatory depression (−14% vs. −41%), than those from non-supplemented control animals. This corresponded to nearly constant cerebral ATP levels in the former versus a 54% decrease in the latter animals after 30 min of anoxia.42 Also, measurements of the maximal respiratory amplitudes in such pups during hypoxia showed an increase by 51%, compared to 22% in control animals.43 A randomized, controlled trial of creatine supplementation (200 mg/kg/d) to preterm infants with AOP, however, starting at around 2 wk of age and being administered for 14 d, showed no effect at all on AOP,44 although this could have also been due to an insufficient dose and/or duration of treatment. This issue deserves further study.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


