Metabolic Acidosis in the Newborn
INTRODUCTION
Metabolic acidosis in the neonate can be caused by several reasons, including increased acid intake from exogenous sources; increased endogenous production of an acid, such as seen in an inborn error of metabolism (IEM); inadequate excretion of acid by the kidneys; or excessive loss of bicarbonate in urine or stool. Presence or absence of an anion gap (AG) can help to distinguish the underlying etiology.
In general, with a pure or uncompensated metabolic acidosis, every 10 mEq/L fall in bicarbonate (HCO3) results in an average pH fall of 0.15. Neonates have an average arterial pH of 7.37 (range of 7.35–7.45). The average bicarbonate level in a neonate is 20 mEq/L. A diagnosis of metabolic acidosis can be made when the pH is less than 7.35 and a base deficit greater than 5 exists.1
The AG is calculated by subtracting the serum concentrations of the measured anions (bicarbonate and chloride) from the cation sodium (Figure 60-1). The AG equation can be written as AG = ([Na+]) – ([Cl–] + [HCO3–]). A normal AG is typically less than 12 mEq/L.1 If the AG is elevated (ie, > 15 mEq/L), then there are anions that have not been accounted for, and an investigation must be performed to search for the culprit. Common anions that result in an elevated AG include lactate and the ketone bodies β-hydroxybutyrate and acetoacetate. Neonates with intoxication IEMs usually have elevated AGs from accumulation of the toxic organic acid, such as isovaleric acid in isovaleric acidemia (IVA), in addition to lactate and ketone bodies secondary to clinical decompensation.

FIGURE 60-1 Anions are molecules that contain a negative charge. Cations are molecules that contain a positive charge. Generally, the total amount of anions and cations in plasma, serum, or urine are equivalent. The anion gap (AG) is the difference between the measured anions and cations and can be calculated by subtracting the concentrations of bicarbonate and chloride from sodium. The AG equation can be written as AG = ([Na+]) – ([Cl–] + [HCO3–]). A normal AG is less than 12 mEq/L. If the AG is elevated, then there are anions in the bodily fluid that have not been accounted for in the equation. Common anions that result in an elevated AG include lactate and the ketone bodies β-hydroxybutyrate and acetoacetate. Organic acids that accumulate in inborn errors of metabolism (IEMs) can also lead to an increased anion gap.
Infants will try to correct metabolic acidosis by a reflex respiratory alkalosis using hyperventilation and Kussmaul respirations. More severe uncompensated acidosis can decrease peripheral vascular resistance and cardiac ventricular function, leading to hypotension, pulmonary edema, and tissue hypoxia, which will further complicate the picture by increasing lactate production because of hypoxia and poor perfusion.
Infants with IEMs are usually normal at birth. They develop nonspecific findings similar to sepsis, such as poor feeding, vomiting, lethargy, hypotonia, hypothermia, seizures, and coma. In any child less than 4 weeks old who presents with sepsis, consider additional testing to look for an IEM. Although expanded newborn screening to look for IEMs is regular practice in the United States, remember that this is just a screen; it is neither comprehensive nor diagnostic. If there is a clinical concern for an IEM, additional testing should be done regardless of the result of the newborn screen.
Clues that increase the likelihood of identifying an IEM include a family history of consanguinity or history of a neonatal or sibling death. Occasionally, maternal history of acute fatty liver of pregnancy (AFLP) may be solicited. In addition, physical findings of Kussmaul respirations, hepatomegaly, or unusual odor may be seen. For example, individuals with untreated IVA can smell like sweaty socks.
Treatment of the metabolic acidosis depends on cause. Although sodium bicarbonate can be given to improve the pH, it will not treat the underlying cause of the acidosis, especially if the underlying etiology is an IEM. The typical dose of sodium bicarbonate is 1 mEq/kg per dose. If too much bicarbonate is given, calcium and potassium levels will fall and can lead to complications like tetany, seizures, and arrhythmias.
CAUSES OF METABOLIC ACIDOSIS WITH NORMAL ANION GAP
Metabolic acidosis with a normal AG is usually the result of intestinal or renal loss of bicarbonate. In this situation, an increase in chloride compensates for the loss of bicarbonate. For this reason, this is also known as hyperchloremic metabolic acidosis.
Intestinal loss of bicarbonate can result from diarrhea, fistulas, or a pancreatic ileostomy. Renal loss of bicarbonate can occur from renal tubular acidosis and renal Fanconi syndrome. Medications, including arginine hydrochloride, which is used for the growth hormone stimulation test, as well as carbonic anhydride inhibitors such as the antiepileptic drug topiramate and the diuretic acetazolamide, can lead to hyperchloremic metabolic acidosis. This type of acidosis can also be seen in individuals with hyperkalemia and those recovering from diabetic ketoacidosis because of shifts in electrolytes and osmolarity.
Renal Fanconi syndrome can be caused by IEMs as well as other causes but does not present with an AG like most of the other IEMs. Infants will have dehydration, failure to thrive, and rickets due to urine loss of phosphate. Diagnostic laboratory investigations include findings of elevation of multiple amino acids on urine amino acids analysis (known as generalized aminoaciduria), glucosuria in the presence of normoglycemia, and hyperphosphaturia.1
Cystinosis, galactosemia, glycogen storage disorders (especially types I and III), hereditary fructose intolerance, tyrosinemia I, Fanconi-Bickel (glycogen storage disorder XI), Wilson disease, Lowe syndrome, mitochondrial disorders, and osteopetrosis can all manifest as renal Fanconi syndrome. Consequently, other testing is necessary to identify the underlying cause.
Medications, including antibiotics such as tetracycline and the aminoglycosides; chemotherapeutic agents such as cisplatin, ifosfamide, 6-mercaptopurine; the common seizure medication valproate, and some human immunodeficiency virus (HIV) medications, including tenofovir, can all lead to renal Fanconi syndrome. Other causes include heavy metal exposure to cadmium, lead, mercury, platinum, and uranium and several Chinese herbs. More commonly seen in adults, the dysproteinemias, including multiple myeloma, amyloidosis, light-chain nephropathy, and benign monoclonal gammopathy, can also lead to renal Fanconi syndrome. Finally, immunologic injury resulting from interstitial nephritis, renal transplantation, or malignancy can be a cause of renal Fanconi syndrome.1
CAUSES OF METABOLIC ACIDOSIS WITH ELEVATED/INCREASED ANION GAP
The differential for metabolic acidosis includes a number of disorders that increase the AG with free acids or decreased buffering by bicarbonate. The memory aid MUDPILES can be used to remember the possible sources that increase the AG (Table 60-1). Although some of the conditions in the MUDPILES list may not be relevant in the neonate, do not forget to consider the maternal contribution to the current clinical state of the neonate, especially if the child is less than 48 hours old or is consuming breast milk.
Table 60-1 Causes of Metabolic Acidosis With Elevated Anion Gap
MUDPILES
Methanol, metformin
Uremia
Diabetic ketoacidosis
Paraldehyde, propylene glycol, phenformin
Iron, isoniazid, inborn errors of metabolism (see separate section this table)
Lactic acid
Ethanol, ethylene glycol (antifreeze)
Salicylates
Ketosis
Diabetic ketoacidosis
Starvation
Inborn errors of metabolism (see separate section this table)
Lactic acidosis
Sepsis
Shock
Tissue hypoxia and poor perfusion
Congenital heart defect
Inborn errors of metabolism (see separate section this table)
Inborn errors of metabolism
Disorders of amino acid metabolism
Propionic acidemia
Methylmalonic acidemia
Isovaleric acidemia
Maple syrup urine disease
Multiple carboxylase deficiency
Betaketothiolase deficiency
Disorders of carbohydrate metabolism
Pyruvate carboxylase deficiency
Pyruvate dehydrogenase deficiency
Fructose-1,6-bisphosphatase deficiency
Glycogen storage disease type I
Disorders of fatty acid metabolism
Very long-chain acyl-CoA dehydrogenase deficiency
Long-chain hydroxyacyl-CoA dehydrogenase deficiency
Carnitine palmitoyltransferase I
Carnitine palmitoyltransferase II and carnitine translocase deficiency
Abbreviation: CoA, coenzyme A.
An important source of free acid in neonates is lactate, which is included in the MUDPILES mnemonic. Lactic acidosis is a common finding in the neonate and can result from primary or secondary causes. Secondary causes are numerous and include seizure, infection, sepsis, shock, hypoxia, anoxia, hypovolemia, hemorrhage, poor perfusion or circulation, congenital heart defects, and the necessity for resuscitation in addition to several of the IEMs.1 If lactic acidosis is identified, testing for secondary causes such as with blood, urine, cerebrospinal fluid (CSF) cultures in addition to a good cardiovascular examination with 4 extremity blood pressures and echocardiogram should be performed. Even if 1 of these secondary causes is identified, additional testing for an IEM should be considered.
In addition to the MUDPILES list, ketosis is a common cause for an increased AG. Although ketosis can be seen during times of illness, when consuming a ketogenic or very-low-calorie diet, or starvation, it is always pathologic in the neonate. Finally, the major component of this chapter is the discussion of IEMs.
Inborn errors of metabolism are disorders in which a particular enzyme is unable to function appropriately in the breakdown of amino acids, carbohydrates, or fatty acid oxidation (Figure 60-2). The enzyme deficiency often leads to accumulation of substrate and insufficiency of product. The precursors that accumulate in IEMs are often acids or are processed into acids that can act as intoxicating molecules and damage organs, including the liver, pancreas, kidney, heart, skeletal muscle, and brain, either directly or by inhibiting the organ’s usual functions, such as ammonia breakdown, or production of energy.
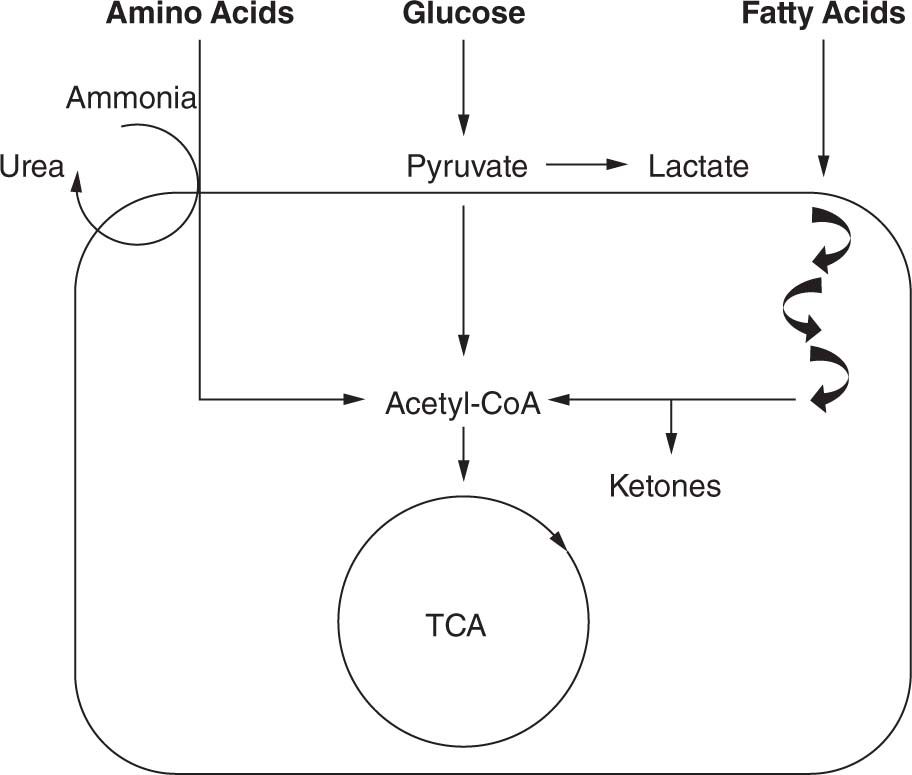
FIGURE 60-2 Basic pathways. Amino acids, carbohydrates, and fatty acids all undergo multiple processes to be converted into energy. The final common pathway is the formation of acetyl-CoA (coenzyme A), which can enter the tricarboxylic acid (TCA) cycle. Hydrogenated nicotinamide adenine dinucleotide (NADH) generated by the TCA cycle can be fed to the electron transport chain to generate adenosine triphosphate (ATP), the main energy source for numerous cellular functions. Defects in amino acid, carbohydrate, or fatty acid metabolism will lead to decreased energy production.
The IEMs that present in the neonatal period include those of the intoxication type, such that they present with decreased feeding and increasing lethargy, which can progress to encephalopathy, seizures, coma, and even death if not treated. One of the major indications of any of these disorders is the presence of metabolic acidosis, especially with an AG because of the accumulation of free acids secondary to the enzyme block. Most of these disorders can be diagnosed by routine metabolic testing (Table 60-2), including plasma amino acid and plasma acylcarnitine profile and urine organic acid analyses.
Table 60-2 Laboratory Tests for Any Patient With a Known or Suspected Inborn Error of Metabolism (IEM)
Venous/arterial blood gas
Glucose
Lactate
Ammonia
Comprehensive Metabolic Panel
Anion gap ([Na+] – ([Cl–] + [HCO3–])
Complete blood cell count
Blood culture
Plasma amino acids
Plasma acylcarnitine profile
Urinalysis to assess ketones
Urine organic acids
Urine culture
SPECIFIC DETAILS ABOUT IEMs THAT PRESENT WITH METABOLIC ACIDOSIS
Disorders of Amino Acid Metabolism
Disorders of amino acid metabolism will result in the accumulation of organic acids and are also referred to as organic acidemias (OAs). The OAs are a series of disorders that result in free organic acids detectable in urine. Many of these disorders are on the newborn screen in the United States, but infants can present with symptoms of metabolic acidosis prior to obtaining the results of the screening test. The following sections do not provide an all-inclusive list of disorders but rather the more common of these disorders (Figure 60-3).
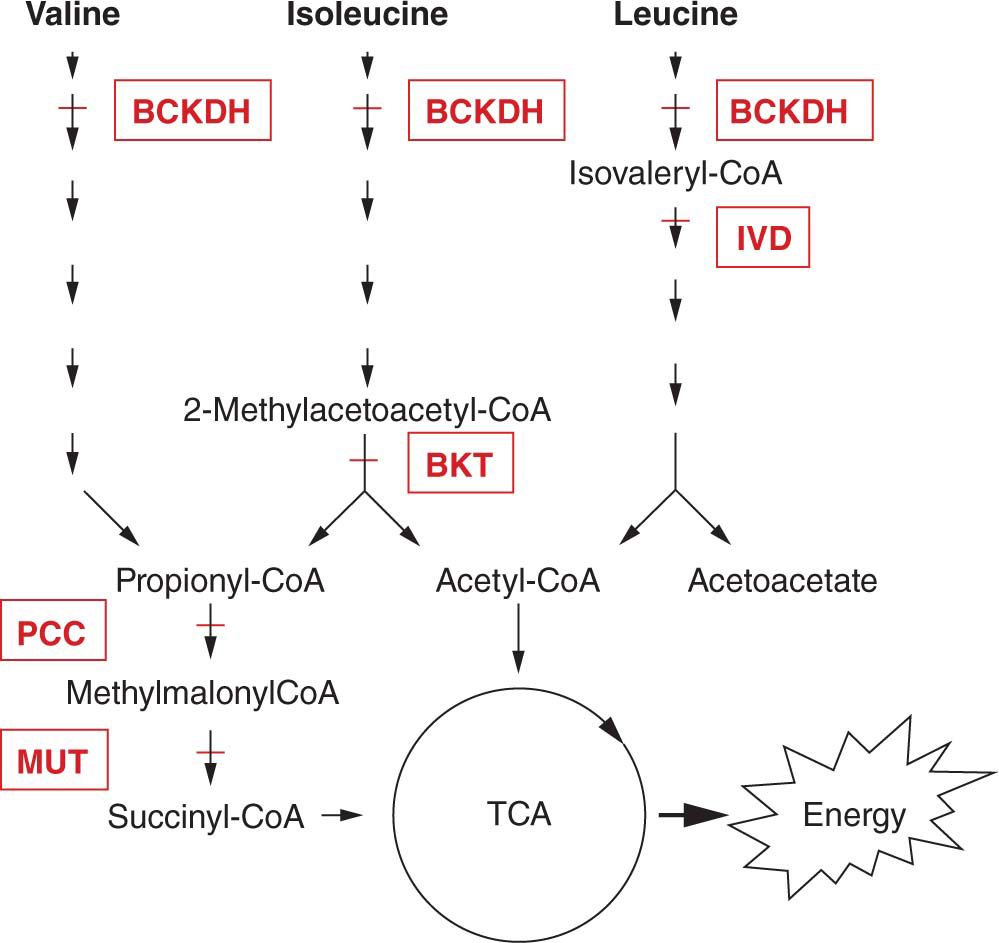
FIGURE 60-3 Detailed pathway of amino acid metabolism. The branched-chain amino acids valine, isoleucine, and leucine share a common enzymatic conversion by branched-chain α-ketoacid dehydrogenase (BCKDH) for the second step of their metabolism; deficiency of BCKDH results in maple syrup urine disease (MSUD). The next step in the leucine pathway is the formation of isovaleryl-CoA (coenzyme A), which is then converted to 3-methyl-crotonyl-CoA by isovaleryl-CoA dehydrogenase (IVD); deficiency of IVD results in isovaleric acidemia. Valine undergoes 5 additional steps, and isoleucine undergoes 4 additional steps prior to the formation of propionyl-CoA. In the isoleucine pathway, just prior to the formation of propionyl-CoA is the conversion of 2-methyl-acetoacetyl-CoA to propionyl-CoA and acetyl-CoA by β-ketothiolase (BKT); deficiency of BKT leads to BKT deficiency. Acetyl-CoA can enter the tricarboxylic acid (TCA) cycle to generate adenosine triphosphate (ATP). Propionyl-CoA is converted to methylmalonyl-CoA by propionyl-CoA carboxylase (PCC); deficiency of PCC leads to propionic acidemia. Methylmalonyl-CoA is converted by methylmalonyl-CoA mutase (MUT) to succinyl-CoA, which can then enter the TCA cycle to generate ATP; deficiency of MUT leads to methylmalonic aciduria.
Propionic Acidemia
Propionic acidemia (PA) is caused by deficiency of propionyl-coenzyme A carboxylase (PCC), which is a biotin-dependent, mitochondrial enzyme with 2 subunits (α and β).3 PA is inherited in an autosomal recessive manner in which mutations in both copies of 1 of the 2 PCC-encoding genes, PCCA at 13q32 (α subunit) or PCCB at 3q21-22 (β subunit), lead to disease. The incidence is approximately 1 in 100,000 newborns worldwide but is more common in Saudi Arabia and the Amish and Mennonite groups. PCC is necessary for breakdown of valine, odd-chain fatty acids, methionine, isoleucine, and threonine (VOMIT). Patients present with poor feeding, vomiting, dehydration, hypotonia, seizures, and lethargy shortly after birth as well as during times of metabolic decompensation, usually with an intercurrent illness or stress. Significant morbidity and mortality are secondary to hyperammonemia, infection, cardiomyopathy, pancreatitis, and basal ganglial stroke.2–5
Laboratory studies in an untreated or metabolically decompensated patient with PA can include evaluation for metabolic acidosis, lactic acidosis, hyperammonemia, ketosis, and hypo- or hyperglycemia. Several cases also presented with pancytopenia, including neutropenia, anemia, and thrombocytopenia. Plasma amino acids show an elevated glycine, which was 1 of the first observations, so PA was initially known as “ketotic hyperglycin(a)emia.”6,7 Urine organic acids show elevations of 3-hydroxypropionate, propionylglycine, tiglylglycine, and methylcitrate. Carnitine can be deficient because of a secondary carnitine deficiency. Acylcarnitine profile analysis shows elevated propionylcarnitine (C3), which is the basis of detection by expanded newborn screening. Confirmation of diagnosis can be done by sequence analysis of PCCA and PCCB as well as PCC enzyme activity on blood or fibroblasts. Prenatally, chorionic villi sampling or amniocentesis can be done to probe for known family mutations, PCC activity, or methylcitrate levels for prenatal diagnosis.
Acute management in a severely affected patient with PA focuses on reversal of catabolism and scavenging of toxic metabolites. A complete protocol developed for PA acute management is available in an article by Chapman et al.4 See Table 60-3 for acute management guidelines and Table 60-4 for discharge planning. Briefly, blood for glucose, electrolytes, ammonia, AG, blood gas, serum urea nitrogen, creatinine, amylase, lipase, and urinalysis for ketones is drawn to determine the level of decompensation. As with most of the intoxication disorders, the initial step of intervention includes the rapid reversal of catabolism, which is often obtained using a 10% dextrose-containing intravenous solution running at least at 6–8 mg glucose/kg/min in a neonate and 20% intralipids (2–3 g/kg/d). Toxic intermediates can be scavenged using levocarnitine (100 mg/kg/d) as well as sodium benzoate/sodium phenylacetate for hyperammonemia prior to confirmation of PA. Infants with severe metabolic acidosis and hyperammonemia do benefit from the rapid reduction of toxic intermediates seen with hemodialysis, extracorporeal membrane oxidation (ECMO), and hemofiltration. Neonates with PA can also have the complication of cardiomyopathy, so an emergent echocardiogram is occasionally useful in the correct setting.
Table 60-3 Acute Management Protocol
Stabilization
Intubate and ventilate, if necessary.
Place intravenous lines. Do not use right intrajugular or 1 of the 2 femoral sites (these locations are necessary for hemodialysis). If unable to obtain access, intraosseus and nasogastric tube are acceptable.
Add vasopressors as necessary to maintain blood pressure.
Normal saline fluid bolus can be given. Avoid overhydration and do not delay reversal of catabolism to give normal saline bolus.
Draw laboratory studies mentioned in Table 60-2; do not discard any blood left over after analysis. If the presenting institution cannot run the laboratory tests, transport samples with the patient.
Reversal of Catabolism
Stop all sources of protein (enteral and parenteral nutrition).
Give nonprotein calories in the form of intravenous fluids with minimum 10% dextrose and electrolytes at 120-150 mL/kg/d or goal of 6–8 mg glucose/kg/min.
If intralipids are available and a fatty acid oxidation defect is not suspected, give 2–3 g/kg/d to provide additional calories.
Do not stop calorie delivery for any reason (eg, medications, additional required fluid bolus, or hyperglycemia).
Consider an insulin drip (0.01 units/kg/h). If hypoglycemia occurs, increase dextrose amount or delivery rate.
Other interventions
Start antibiotics after blood cultures are drawn.
If newborn screen results are unknown, call state laboratory to check for results or expedite their processing (normal results do not rule out metabolic disease).
Arrange transport to a metabolic center as soon as there are concerns for metabolic disease or decompensation.
Arrange for and initiate hemodialysis, hemofiltration, or extracorporeal membrane oxygenation (ECMO) if clinical or laboratory evaluations identify ammonia greater than 300 μmol/L, extreme acidosis/electrolyte imbalance, coma, dilated pupils, poor neurological findings, or increased respiratory rate and based on clinical judgment. Do not use peritoneal dialysis.
For hyperammonemia in an undiagnosed patient, consider starting sodium benzoate/sodium phenylacetate (at same doses as for urea cycle defects).
Intravenous carnitine 100 mg/kg/dose 3–4 times daily over a minimum of 30 minutes can be given if urine output is appropriate (or hemofiltration ongoing) and there is no concern for a long-chain fatty acid oxidation defect.
Follow ammonia, electrolyte, and blood gas values at regular intervals. The frequency is dictated by the patient’s condition and the speed at which results can be obtained.
Protein should be reintroduced within 24–36 hours of initiation of therapy. Protein must be reintroduced even if patient is on hemodialysis or ECMO.
Consider sedation, ventilation, and chemical paralysis if aggressive management is necessary. If used, continuous electroencephalography (EEG) is helpful to monitor progress.
Transfuse packed red blood cells as necessary based on age, clinical condition, and physician discretion.
Transition from acute to chronic management and discharge planning
Establish home metabolic nutrition support regimen.
Provide gastrostomy tube (G tube) or nasogastric tube (NG tube) placement for enteral feeding.
Medications should be transitioned to oral administration.
Complete studies including echocardiogram, electrocardiogram (ECG), hearing screen, optic fields (if cerebral edema) and dilated ophthalmologic examination according to underlying diagnosis.
Involve physical therapy and occupational therapy and refer to early intervention.
Any parental training needed is completed prior to discharge.
Table 60-4 Discharge instructions
Parental training
Preparation of metabolic formula with a home gram scale
G-tube or NG-tube (if present) care
Lessons in giving all medications
Instructions on how to use ketosticks
Medications and formula
Prescriptions for all medications and ketosticks
Give at least 2 days prior to discharge to have time to fill
One-week supply of all medications
List of medications with dosages, times, and reasons for use
Several cans of metabolic formula
Paper and electronic copy of diet with list of feeding times
Follow-up
Emergency room letter
Emergency contact information for the metabolic physician and clinic
Appointment date and time for metabolic clinic and other specialists
Referral to early intervention
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


