Mechanisms of Hormone Action
Adda Grimberg and Linda A. DiMeglio
A hormone is a molecule that is secreted into the circulation to influence the function of target cells in another tissue or organ. In addition to distant targets via the circulation (endocrine action), many hormones also influence the function of the cells that secreted them (autocrine action) or that of adjacent cells (paracrine action). 
Hormone actions are mediated via systems with similar basic components. This chapter will describe the specific components of the major categories of hormones and will provide a methodological overview of how these hormones contribute to health and disease. Each system is characterized by the gland that produces a hormone, the hormone itself, and often circulating binding proteins. The binding proteins create a circulating pool of readily available hormone stores, alter hormone clearance and distribution, and modulate hormone activity rates. Binding of the free hormone, the ligand, to specific receptors on the target cells initiates a variety of signal transduction pathways, often through a series of signaling cascades or second messengers that alter cell function. The impact of the hormonal message on cell function is often modified by other regulatory molecules that modulate the activity of these signaling pathways.
HORMONE CLASSIFICATION
Hormones are categorized based on their molecular structure. Most are peptide hormones, made of amino acid chains. Small neuropeptide hormones include antidiuretic hormone (ADH), gonadotropin-releasing hormone (GnRH), and thyrotropin-releasing hormone (TRH). Larger peptide hormones, known as protein hormones, include insulin and growth hormone (GH). Glycoprotein hormones have carbohydrate side chains attached; these include human chorionic gonadotropin (hCG), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH). Amino acid–derived peptide hormones have an NH2 group at the end of the molecule and arise from the amino acids tyrosine and tryptophan. These include thyroxine, dopamine, catecholamines, and melatonin. In general, peptide hormones work through cell surface receptors.
In contrast, steroid hormones are lipid and phospholipid derivatives. These hormones are synthesized from cholesterol (eg, testosterone, cortisol) or the eicosanoids (prostaglandins) by a series of enzymatic steps. Other hormones are vitamin derivatives, including the retinoids (vitamin A) and vitamin D. In general, thyroid hormones, steroid hormones, retinoids, and vitamin D are lipid-soluble and work through intracellular receptors.
HORMONE RECEPTOR CLASSIFICATION
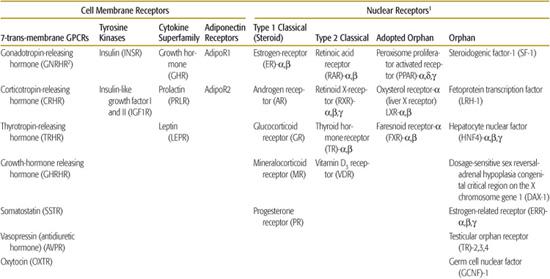
Hormone receptors form two major categories, cell surface or cytosolic nuclear, based on their cellular localization. These are further subdivided according to their structure and signaling mechanisms (Table 520-1).2 Because peptide hormones cannot penetrate the cell membrane, they bind to receptors located on the cell surface. Activation of such cell membrane receptors initiates signaling cascades that involve second messenger systems to effect changes within the cell. There are four main classes of cell membrane receptors: G-protein-coupled receptors, tyrosine kinase receptors, cytokine superfamily receptors, and adiponectin receptors. In contrast, lipophilic hormones have cytosolic receptors, which translocate to the nucleus when bound to ligand. In the nucleus, the activated receptors modify transcription of target genes by DNA-specific binding to “response elements” in the promoters of those genes. Broadly, nuclear receptors are classified into ligand-activated and “orphan” receptors (those with no known endogenous ligand). The ligand-activated receptors include “classical” receptors (for steroid, thyroid, and retinoid hormones) and “adopted orphan” receptors (fatty acid, cholesterol/bile acid, and xenobiotic), which were originally identified because they were targets of exogenous substances but were subsequently found to have endogenous ligands as well.
 CELL MEMBRANE RECEPTORS
CELL MEMBRANE RECEPTORS
G-Protein-Coupled Receptors
Seven-transmembrane (or heptahelical) G-protein-coupled receptors (GPCRs) are named after their characteristic structure and their common use of heterotrimeric G proteins to couple to second messenger pathways. GPCRs comprise the largest class of receptors, close to 1000 in number.3 As described in this chapter, these receptors are used by hormones (Table 520-1), but they are also employed by the sensory apparatus (such as rhodopsin receptors in vision and odorant receptors in olfaction), and some have endogenous ligands that remain unknown (“orphan” receptors). 
GPCRs all contain an extracellular ligand-binding domain, seven transmembrane alpha-helices arranged in a bundle, and a carboxy-terminal cytoplasmic tail. G proteins are heterotrimeric and contain an alpha, a beta, and a gamma subunit, which serve as the molecular switch between GPCRs and their second messenger systems, as shown in Figure 520-1. 
Table 520-1. Classes of Hormone Receptors and Their Ligands

Agonist binding activates GPCRs within milliseconds, inducing distinct active conformations of the GPCR that in turn activate G proteins, the rate-limiting step in GPCR signaling.11 Allosteric binding at sites distinct from the agonist-binding site may also induce conformational changes that alter GPCR activity,12 and some GPCRs have constitutive activity independent of ligand binding. Abnormalities of ligand-binding, mutations that lead to an alteration in GPCR constitutive activity, or those that impinge upon the intracellular transport and expression of functional surface receptors (oligermerization defects that trap the GPCR in the endoplasmic reticulum) all may cause functional disorders. For example, short stature is associated with a mutation of the ghrelin receptor that disrupts the receptor’s constitutive activity, but not its binding, and activation by ghrelin.13 Many toxic thyroid nodules (“hot thyroid nodules”) appear to be due to activating mutations in the TSH receptor (TSHR).14 Similarly, activating mutations in the LH receptor lead to precocious puberty in males.15,16
One mode of signaling stimulated by GPCRs is via adenylyl cyclase/cyclic adenosine monophosphate (cAMP) mechanisms. G-alpha S activates all isoforms of adenylyl cyclase, a family of enzymes comprised of two cytosolic and two transmembrane domains that synthesize cAMP from ATP (Fig. 520-1A).  cAMP’s best studied effector is protein kinase A (PKA), a holoenzyme that phosphorylates serine or threonine of specific substrates. A-kinase anchoring proteins (AKAPs) serve as scaffolding for PKA-interacting proteins and target PKA to specific substrates and subcellular compartments.21 One of the classic PKA substrates is the transcription factor cAMP response element-binding (CREB) protein.22 PKA activity also regulates cation channel activity. In addition to PKA, cAMP signals through cAMP-gated ion channels and through the guanine nucleotide exchange factor EPAC (exchange protein activated directly by cAMP),20 which activates Rap, a GTPase of the Ras family that is transduced to effectors such as integrins, gap junctions, and phospholipase C. EPAC also mediates cAMP-regulated ion channel activity, intracellular calcium (Ca+2) signaling, and exocytosis.20,23
cAMP’s best studied effector is protein kinase A (PKA), a holoenzyme that phosphorylates serine or threonine of specific substrates. A-kinase anchoring proteins (AKAPs) serve as scaffolding for PKA-interacting proteins and target PKA to specific substrates and subcellular compartments.21 One of the classic PKA substrates is the transcription factor cAMP response element-binding (CREB) protein.22 PKA activity also regulates cation channel activity. In addition to PKA, cAMP signals through cAMP-gated ion channels and through the guanine nucleotide exchange factor EPAC (exchange protein activated directly by cAMP),20 which activates Rap, a GTPase of the Ras family that is transduced to effectors such as integrins, gap junctions, and phospholipase C. EPAC also mediates cAMP-regulated ion channel activity, intracellular calcium (Ca+2) signaling, and exocytosis.20,23
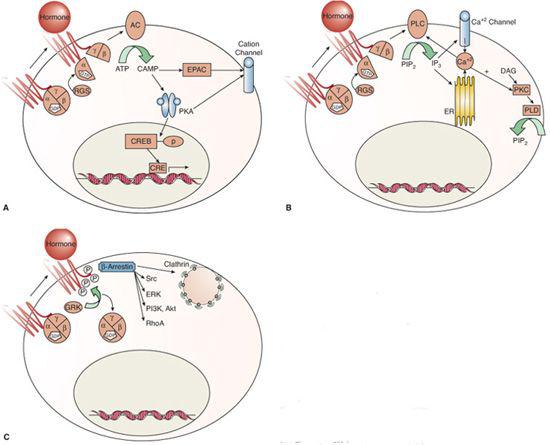
FIGURE 520-1. Signaling mechanisms of G protein–coupled receptors. Binding of hormone leads to conformational changes in the G-protein coupled receptor (GPCR) with G protein–dependent (A and B) and independent (C) mechanisms of action. Ligand (hormone) binding to the GPCR can activate its guanine nucleotide exchange function, which exchanges guanosine triphosphate (GTP) for the guanosine diphospate (GDP) that is bound to the G-alpha subunit in its inactive state.5,6 GTP binding to G alpha induces a structural rearrangement whereby G alpha GTP and G beta/gamma dissociate from the GPCR and interact with effectors of the second messenger systems.6 The signal terminates with hydrolysis of GTP to GDP, which allows G alpha GDP to reform an inactive heterotrimer with G beta/gamma. This GTPase activity is catalyzed by the G-alpha subunit, which is accelerated by regulator of G-protein signaling (RGS) proteins that bind to the activated G-alpha subunits.7 Subsequent signaling activity is mediated via cAMP pathways (A) or via alterations in phospholipids and calcium fluxes (B). Hormone binding may also stimulate G protein-independent activity (C).
GPCRs also act via activation of phospholipases, as shown in Figure 520-1B. Phospholipids are the most abundant of cell membrane lipids and are characterized by a polar head group, a phosphate group, a glycerol backbone, and two fatty acid tails.24 Both cell membrane and intracellular membrane phospholipids are cleaved by phospholipases; phospholipase C cleaves off the polar head phosphate (leaving a diacylglycerol [DAG] molecule), phospholipase D cleaves off the polar head group (leaving the phosphate and fatty acids on the glycerol backbone), and phospholipase A cleaves off one free fatty acid (leaving a lysophospholipid molecule). Each of the products serves in signal transduction, frequently in interconnected pathways.
Phospholipase C occurs in six families containing at least 13 isoforms.25,26 Phospholipase C (PLC) activity creates DAG, an integral membrane component, and stimulates signaling via protein kinase C (PKC) and chimaerins (Rac GTPase-activating proteins).24 The most important of the many phospholipase C substrates is phosphatidylinositol(4,5)-bisphosphate [PI(4,5)P2], with the resultant product being PI-(1,4,5)-triphosphate [IP3], which leads to calcium fluxes, as shown in Figure 520-1B.
Following GPCR activation, intracellular Ca+2fluxes can result from cAMP and phospholipase signaling. Phospholipase C-released IP3 binds to intracellular receptors in the endoplasmic reticulum (ER), rapidly mobilizing Ca+2 from the ER stores; this is followed by a slower Ca+2 influx from outside the cell via voltage-gated Ca+2 channels.27 The rise in the intracellular Ca+2 concentration activates enzymes for further signaling. The combined regulation of Ca+2 fluxes by lipid signaling and the Ca+2 dependence of many enzymes in lipid signaling fosters amplification of GPCR downstream signaling.
Hormone binding to the G-protein receptor may also activate other kinases known as G-protein receptor kinases (GRKs), which are independent of the classical GPCR kinases, as shown in Figure 520-1C. Seven GRKs have been identified to date.8 These GRKs phosphorylate the cytoplasmic tail and some intracellular domains of the agonist-bound GPCR, thereby recruiting beta-arrestins. Beta-arrestins block further interaction between the GPCR and the G proteins (they become uncoupled) and target the ligand-bound GPCRs for endocytic internalization, thereby terminating the signal and preventing further GPCR stimulation.28 This mechanism underlies the phenomenon of agonist-induced desensitization of GPCRs.29 However, there is growing appreciation that beta-arrestins can also provide direct signaling functions independent of the G proteins.30
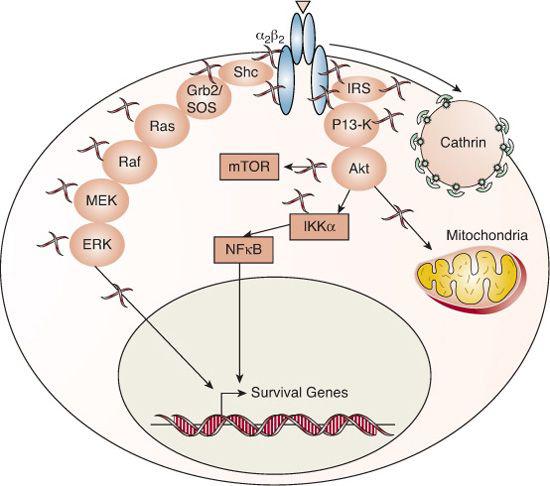
FIGURE 520-2. Signaling mechanisms of tyrosine kinase receptors. The insulin and IGF receptors are composed of two alpha subunits that participate in extracellular hormone binding and two beta subunits that traverse the cell membrane and possess intrinsic tyrosine kinase activity in their cytoplasmic termini. Binding of hormone to the receptor activates its autophosphorylation function; the phosphorylated tyrosine residues act as docking sites for adapter molecules like the SRC homology 2 domain-containing protein (SHC) and the insulin receptor substrate 1 (IRS1) family. The adapter molecules recruit and initiate the phosphorylation cascades of MAP kinase and PI3K, leading to extracellular-regulated protein kinase (ERK), Akt (Protein kinase B), and mammalian target of rapamycin (mTOR) activation. Ultimately, the tyrosine-kinase receptors transduce signaling to the nucleus (such as NF-kappa-B translocation and induction of c-myc and c-Fos) and mitochondria to elicit survival, mitogenic and metabolic activities of the cell.35 Insulin and IGF1 receptors are internalized in the ligand-bound, active state by the endocytic machinery into clathrin-coated pits.
Tyrosine-Kinase Receptors
About 60 receptors from a score of subfamilies belong to the tyrosine-kinase receptor superfamily. All share a ligand-binding extracellular domain and an intracellular tyrosine-kinase domain. The tyrosine-kinase receptors of many growth factors, including epidermal growth factor (EGF), fibroblast growth factor (FGF), and nerve growth factor (NGF), are single transmembrane polypep-tides that dimerize or oligomerize upon ligand binding.33 In contrast, the receptors for the hormones insulin, IGF-I and IGF-II, are covalent dimers linked by disulfide bonds.34 Binding of hormone to the receptor activates its autophosphorylation function, as shown in Figure 520-2.
Both IGF-I and IGF-II signal via the type 1 IGF receptor (IGF1R); the IGF-2 receptor is the mannose-6-P receptor, a large, single-transmembrane receptor that serves as a clearance binding protein for IGF-II.36 The IGF1 receptor shares over 50% amino acid sequence homology with the insulin receptor and 84% homology within their tyrosine-kinase domains.37 The insulin receptor exists in two isoforms, A and B, created by alternative splicing of exon 11 of the INSR gene that leads to a 12-residue difference in the insulin receptor alpha subunit.38 While IGF-1 receptor and the insulin receptor have high-affinity binding for their cognate hormone ligands, they also show low-affinity binding for the converse hormones due to the homologous nature of the receptors and ligands; however, insulin receptor-A binds the IGFs with high affinity as well.34 Also owing to the homology of the system, IGF-1 receptor and insulin receptor hemireceptors can join in mixed fashion to form functional hybrid receptors. Thus, the insulin and IGF systems possess great potential for cross talk. Specificity is conferred by the relative bioavailability of the hormones, the relative expression of the various receptors (expression of the insulin receptor isoforms is tissue- and development-dependent), the differences in binding affinities, the different abilities to activate downstream pathways, and the distinct spatial/temporal compartmentalization of the signaling via receptor trafficking.39-43
Cytokine Receptor Superfamily, Class I
The GH receptor (GHR), prolactin receptor (PRLR), and leptin receptor (LR) are among the over 40 members of the class I cytokine receptor superfamily. These receptors activate associated kinases but do not possess intrinsic catalytic activity themselves. They occur in both short and long forms. The GH receptor is cleaved to release its extracellular domain, which forms the soluble, circulating GH-binding protein (GHBP).44 In contrast, the short form of the prolactin receptor is a membrane-bound receptor.45 There are at least five isoforms of the leptin receptor produced by alternative splicing from the OBR gene.46
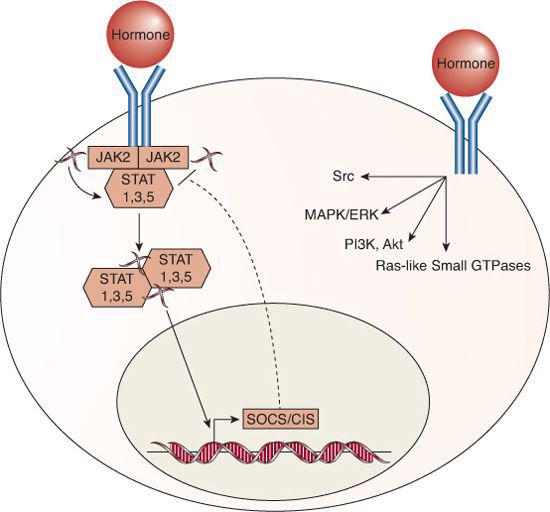
The long forms of the GH, prolactin, and leptin receptors are all transmembrane proteins that recruit the tyrosine kinase Janus kinase 2 (JAK2); this, in turn, activates the signal transducers and activators of transcription (STATs) that function as transcription factors that stimulate and repress transcription of multiple target genes, as shown in Figure 520-3.46-49 Additional signaling pathways are also activated by each of the receptors. For example, apart from JAK2, the GH receptor can directly activate the Src tyrosine kinase,48 and GH, prolactin, and leptin receptors activate the MAP kinase pathway, PI3K/Akt, Ras-like small GTPases, and other indirect second messengers.46-48,50
Suppressors of cytokine signaling (SOCS)/cytokine-inducible SH2-containing protein (CIS) family proteins are important targets of the cytokine receptor superfamily, because they form an internal negative feedback loop; expression of the various SOCS/CIS proteins are induced by GH, prolactin, and leptin receptor activity and, in turn, inhibit STAT signaling, thereby attenuating or terminating the hormone action.53-57 Because the cytokine receptor family members signal via phosphorylation cascades, they can also be inhibited by protein tyrosine phosphatases, including SHP1 and SHP2, similar to the tyrosine-kinase receptors.48,54
Adiponectin Receptors
The two adiponectin receptors, AdipoR1 and AdipoR2, are encoded by distinct genes but share 67% amino acid identity.58 They are integral membrane proteins that contain evolutionarily conserved 7-transmembrane domains. AdipoR1/R2 have the opposite topology to that of GPCRs (the N-terminus is internal and the C-terminus is extracellular) and do not signal via G proteins (Fig. 520-4). Therefore, they constitute a separate, new category of cell membrane receptors.
Adiponectin is an adipokine (adipose tissue–derived hormone that helps regulate energy homeostasis) that structurally belongs to the complement 1q family.60 Adiponectin can exist full length (as predominantly found in the plasma) or as a smaller, globular fragment created by proteolytic cleavage. Obesity decreases both plasma adiponectin levels and peripheral expression of AdipoR1/R2, which leads to insulin resistance.60 AdipoR1 expression in the arcuate nucleus of the hypothalamus increases during fasting (decreases with refeeding), activates AMPK, decreases energy expenditure, and stimulates food intake.62
 INTRACELLULAR/NUCLEAR RECEPTORS
INTRACELLULAR/NUCLEAR RECEPTORS
Nuclear receptors elicit biological responses by working as ligand-activated, DNA-binding transcription factors. Unlike cell membrane receptors, which first sense a ligand’s presence or absence and then use complex pathways to send information to a cell, nuclear receptors interact with regions close to gene promoters and regulate specific target gene expression. 
Over 100 nuclear receptors have been described, and a large number of coactivators and corepressors that enhance or decrease the transcription rates of their associated receptors have also been identified.64 Nuclear receptors are classically divided into families based upon their hormone response element recognition, including steroid, retinoid, thyroid, and lipophilic hormones. Phylogenetic analyses have led to nuclear receptors being divided into type I “steroid” receptors (including those for estrogens, progestins, androgens, glucocorticoids, and mineralocorticoids) and type II receptors for all-trans retinoic acid, 9-cis retinoic acid, thyroid hormone, and vitamin D3. Another class of nuclear receptors is the “adopted orphans” that were formerly orphan receptors and are now with recognized binding affinity for an endogenous substance. For example, the peroxisome-proliferator-activator receptor (PPAR-α)65 was first recognized to be the receptor for the cholesterol-lowering fibrate drugs and was subsequently found to bind fatty acids as its endogenous ligand. A final group of nuclear receptors remains “orphan” transcription factors, without a characterized ligand, including steroidogenic factor-1 (SF-1), a key regulator of adrenal steroidogenesis; hepatocyte nuclear factor-4 (HNF-4α), which influences pancreatic beta-cell function; and the dosage-sensitive sex-reversal adrenal hypoplasia congenital critical region on the X chromosome (DAX-1).
Nuclear receptors are highly variable in size, with molecular weights from 46 to 100 KDa. They have a modular structure consisting of five regions, an N-terminal regulatory domain, a DNA-binding domain, a hinge segment, a ligand-binding domain, and a C-terminal domain. The N-terminal domain regulates gene transcription activation. Its sequence and structure vary greatly between nuclear receptors. In some receptors, the N-terminal domain contains an AF1 (activation function) domain. AF1 transcriptional activation is normally weak, but AF1 can synergize with AF2, found in the ligand-binding domain, for more robust upregulation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


