Lysosomal Storage Disorders
Irina A. Anselm
Lysosomal storage disorders (LSDs) comprise a group of approximately 30 relatively rare inherited diseases that are characterized by defective functioning of one or several lysosomal enzymes. As a result of the enzyme deficiency, a storage material (glycogen, glycoproteins, shingolipids, cholesterol) accumulates in various organs, including central nervous system (CNS), liver, spleen, and kidneys. While most individual LSDs are quite rare, as a group their incidence is estimated to be between 1:5000 to 1:10,000. The prevalence of some of LSDs is higher in certain populations. LSDs are classified based on the type of the enzymatic defect or type of stored substrate product. Clinical symptoms (both somatic and neurological manifestations) tend to progress as the substrate accumulates over time. Several LSDs with CNS involvement are discussed below. These disorders are further discussed in Chapters 160 and 161. Diagnosis is usually made by enzyme assay or mutation analysis. Hematopoietic stem cell transplant (HSCT) has been used for the treatment of several LSDs over the past three decades. Enzyme replacement therapy (ERT) became available for the treatment of few LSDs in the past 15 years. New therapies have emerged in most recent years; small molecule drugs (chaperones) can reduce substrate production (substrate reduction therapy) or increase residual enzyme activity (enzyme enhancing therapy). Gene therapy may become available for the treatment of some LSDs in the future and is under investigation.
LYSOSOMAL DISORDERS PRIMARILY AFFECTING GRAY MATTER
LYSOSOMAL STORAGE DISEASES CONTAINING LIPID
 GM1 GANGLIOSIDOSIS
GM1 GANGLIOSIDOSIS
GM1 gangliosidosis is an autosomal recessive disorder caused by deficiency of the lysosomal enzyme, GM1 ganglioside β-galactosidase, a lysosomal enzyme requiring a complex of sphingolipid activator protein (saposin B), protective protein, and neuraminidase enzyme in order to function properly. β-galactosidase deficiency results in accumulation of GM1 ganglioside within the brain and storage of galactose-containing glycoproteins and keratan sulfate within various body organs.
Deficiency of β-galactosidase is expressed clinically as 2 different diseases: GM1 gangliosidosis and Morquio B disease. GM1 gangliosidosis is characterized by a neurodegenerative process with associated visceral involvement. Generalized bone disease without CNS involvement is the hallmark of Morquio B disease. Molecular analysis has confirmed allelic mutations of the same gene in these diseases mapped to chromosome 3 (3p21.33).
The clinical phenotype of GM1 gangliosidosis can be divided into 3 forms: type I (infantile), type II (juvenile), and type III (adult). Type I, or infantile, form presents at birth or during early infancy with characteristic facial appearance of depressed nasal bridge, frontal bossing, epicanthal folds, puffy eyelids, gingival hypertrophy, enlarged tongue, low-set ears, and small jaw. Peripheral and facial edema may be seen. Other features include skeletal dysplasia, manifesting as thoracolumbar kyphoscoliosis and hepatosplenomegaly. Vision is poor; a cherry-red macula develops after several months in at least 50% of children. MRI may show progressive atrophy and delayed myelination. Peripheral blood smear analysis frequently shows vacuolated lymphocytes. Patients become vegetative and die within a few years after onset. Type II, or juvenile-onset, disease has onset usually between 1 and 2 years; symptoms consist of mild bony changes without dysmorphic features. The disease primarily affects the CNS and manifests as gradual developmental delay, ataxia, and chore-oathetoid movements. The first symptom is usually difficulty with walking, followed by loss of speech. A retinal cherry-red spot is usually not present. There is no hepatosplenomegaly. MRI is usually normal, and EEG may be diffusely slow but not diagnostic. The course is protracted, with death usually occurring within the first decade. Type III adult-onset disease is characterized by late-onset neurologic symptoms. 
Morquio disease type B is also associated with deficiency of β-galactosidase. Patients present with generalized skeletal dysplasia without CNS involvement. Despite the enzymatic defect, the phenotype resembles systemic manifestations of other mucopolysaccharidoses.
Galactosialidosis is a complicated variant of GM1 gangliosidosis involving deficiency not only of GM1 ganglioside β-galactosidase, but also of neuraminidase. The biochemical defect is caused by a deficiency of the protective protein, cathepsin A. The gene for cathepsin A is located on chromosome 20, and several mutations have been identified in this disease. Clinical presentation is frequently a severe infantile onset with dysmorphic and neurologic features similar to infantile GM1 gangliosidosis. Many milder variants have been described, making the phenotype quite heterogeneous.
 GM2 GANGLIOSIDOSES
GM2 GANGLIOSIDOSES
The GM2 gangliosidoses are a collection of lysosomal diseases associated with the accumulation of GM2 ganglioside in the lysosomes, predominantly of nervous system cells. The diseases may be caused by a deficiency of β-hexosaminidase A, β-hexosaminidase A and B, or GM2 ganglioside activator protein. Two isoenzymes of β-hexosaminidase exist: Hex A (structure αβ) and Hex B (structure ββ). Tay-Sachs variant is caused by defects in the α-subunit of the enzyme and is associated with deficiency of Hex A and normal activity of Hex B. Sandhoff disease is caused by mutations in the β-subunit gene that result in deficiency of both Hex A and B. The genes encoding the β-hexosaminidase α-subunit (HEXA gene) and β-subunit (HEXB gene) are similar in structure, with nearly 60% nucleotide and amino acid homology, but have been mapped on 2 separate chromosomes, with HEXA localized to 15q23-24 and HEXB to 5q11.2-13.3. The GM2 ganglioside activator protein was mapped to chromosome 5. All 3 genes have been cloned and mutations identified.
Tay-Sachs Disease (β-Hexosaminidase α-Subunit Deficiency)
The infantile onset of β-hexosaminidase α-subunit deficiency (Tay-Sachs disease) begins within the first few months of life. The first symptom is an excessive startle in response to noise, tactile stimuli, or light flashes. This startle response differs from the Moro response of normal infants and consists of quick extension of the arm and legs, frequently with clonic movements, and does not attenuate with stimulus repetition. As the disease progresses, motor development slows and previously acquired skills are frequently lost, along with decreased vocalizations and loss of awareness of the environment. A macular cherry-red spot (Fig. 574-1) occurs in over 90% of infants. Storage of lipids within the retinal ganglion cells causes whitish discoloration of most of the retina except for the fovea, which shows the normal red color. Macrocephaly becomes apparent by the second year. Affected children may develop seizures that can be induced by auditory stimuli. Systemic organs are spared in this form of the disease. A vegetative state develops between age 2 and 3 years and is followed by death in a few years, usually from infection. A late-onset (juvenile/adult) variant of GM2 gangliosidosis, with indolent clinical presentation, has been described. Diagnosis of Tay-Sachs disease (TSD) relies upon the demonstration of absent or extremely low activity of Hex A in white blood cells or other tissues. Before community-based carrier-screening programs were developed in the Ashkenazi Jewish population, the incidence of TSD was about 1 per 3600 births of infants of this descent. The carrier rate for TSD among Jewish Americans of Ashkenazi extraction is about 1 in 30. Extensive counseling and carrier-screening programs reduced the incidence of TSD by 90%. 
Sandhoff Disease (β-Hexosaminidase β-Subunit Deficiency)
The age of onset, duration, neurologic symptoms, and ophthalmologic signs in patients with Sandhoff disease are identical to those seen in TSD, but mild hepatosplenomegaly (secondary to storage of globoside) and bony deformities may rarely be present. Deficiencies of both hexosaminidase A and B are present. A juvenile-onset variant occurs after age 1 year, beginning with clumsiness and gait ataxia, later dystonic posturing and seizures, but no cherry-red macula. Adult-onset patients follow a similar clinical course to chronic late-onset adult forms of Hex A deficiency.
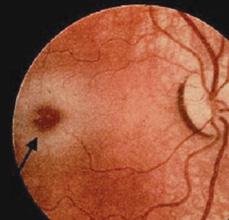
FIGURE 574-1. Cherry-red macula noted in patient with Tay-Sachs disease.
GM2-Activator Deficiency
Individuals with GM2-activator deficiency have disease course similar to those with infantile-onset TSD or B variant. The storage of glycolipids and the pathologic features are also identical to that found in infantile TSD.
MRI in GM2 gangliosidoses demonstrates atrophy with widening of cerebral sulci, increase in ventricular size, and hyperintensities in the basal ganglia, thalamus, and cerebral cortex.  Treatment is limited to symptomatic and supportive care. Attempts at enzyme replacement therapy were unsuccessful, owing to problems with the delivery of the enzyme to the CNS. The experience in treating patients with bone marrow transplantation is very limited.
Treatment is limited to symptomatic and supportive care. Attempts at enzyme replacement therapy were unsuccessful, owing to problems with the delivery of the enzyme to the CNS. The experience in treating patients with bone marrow transplantation is very limited.
 FABRY DISEASE
FABRY DISEASE
Fabry disease is an X-linked condition resulting in lysosomal accumulation of various glycosphingolipids with terminal α-galactosyl residues in the cells throughout the body. It is caused by deficiency of the enzyme α-galactosidase A. In affected hemizygous males, enzyme activity is less than 1% of normal.
Clinical Presentation
Clinical symptoms begin in late childhood or adolescence with periodic crises of pain in the extremities, vascular cutaneous lesions (angiokeratomas), hypohydrosis, corneal and lenticular opacities, strokes, left ventricular hypertrophy, and renal insufficiency. The gene encoding α-galactosidase A (GLA) is localized to Xq22.1. The incidence of Fabry disease has been estimated at 1 in 55,000 male births.
The most debilitating symptom of Fabry disease is pain in the extremities of 2 types: painful crisis and constant discomfort. The painful crises often begin in childhood or adolescence, last from minutes to several days, and may be accompanied by fever and increased erythrocyte sedimentation rate. They are described as intense burning or lancinating pain felt initially in palms and soles, but frequently radiating to proximal extremities and other body parts. In addition to intermittent crises, many patients complain of mild persistent numbness and paresthesias in hands and feet (acroparesthesia). Autonomic nervous system dysfunction manifests as hypohydrosis and anhydrosis (occasionally hyperhidrosis), constipation, chronic diarrhea, and nausea.
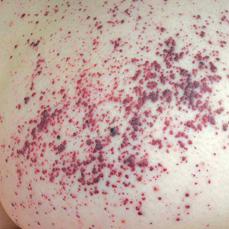
Cerebrovascular manifestations include hemipareses, vertigo, diplopia, dysarthria, nystagmus, headache, ataxia, memory loss, and hemisensory loss. Angiography may reveal tortuosity of brain vessels. Cardiovascular manifestations consist of left ventricular hypertrophy due to the deposition of storage material, mitral valve insufficiency, arrhythmias, and electro cardiographic changes. Progressive glycosphingolipid accumulation in the kidney results in the development of azotemia and renal insufficiency. Urinary sediment contains casts and “Maltese crosses,” which are birefringent lipid globules. Gradual deterioration of renal function leads to death in the third to fifth decade unless treatment with chronic hemodialysis or renal transplantation is provided. Angiokeratomas appear as clusters of ectatic blood vessels, dark-blue or black-blue, in the superficial layers of the skin. These lesions are flat or slightly raised and do not blanch with pressure. Usually, they are located in the groin, buttocks, upper legs, and umbilical region (Fig. 574-2). They appear in childhood and gradually increase in number and size over the years. Corneal opacity, which can be seen only by slit-lamp examination, is usually the first ocular abnormality and is present in essentially all hemizygous Fabry patients and most heterozygous females. Lenticular opacities may occur in approximately 30% of affected males and consist of granular anterior capsular and subcapsular deposits or a characteristic posterior capsular opacity (Fabry cataracts). The corneal and lenticular opacities do not interfere with vision. Heterozygous females may have mild symptoms but sometimes can have severe manifestations, similar to affected males, related to a nonrandom chromosome X inactivation. In approximately 30% of females, a few angiokeratomas may be present in the characteristic locations, and intermittent pain and paresthesias may occur; rarely, cardiac and renal symptoms develop.
MRI shows hyperintensity in the periventricular white matter early in the disease and development of subcortical and cortical strokes over time. There is also a characteristic “pulvinar sign” seen in many male patients, which consists of an increased signal intensity on T1-weighted MRI images in the pulvinar nucleus of the thalamus.1 Nerve conduction studies reveal an elevated threshold to current perception but no changes in nerve conduction velocities.
Diagnosis and Treatment
Diagnosis is confirmed by low activity of α-galactosidase A in leukocytes in affected males. In females, measurement of enzyme activity is unreliable, as many carrier females have normal levels. GLA is the only gene known to be associated with Fabry disease, with almost 100% of affected males having an identifiable mutation. Molecular genetic testing is the only reliable method that should be used to identify affected females.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


