Fig. 9.1
Typically posterior fossa juvenile pilocytic astrocytomas
Within the brainstem an exophytic tumor component is suggestive of a low grade glioma (Fig. 9.2).
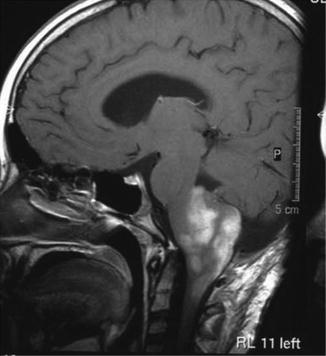
Fig. 9.2
Exophytic brainstem low grade glioma. Reprinted from Warmuth-Metz M. Neoplasms, Brain, Posterior Fossa, Pediatric. In: Baert AL (ed). Encyclopedia of Diagnostic Imaging. Berlin, Germany: Springer-Verlag; 2008:1231–1237. With permission from Springer Verlag
For optic pathway glioma, classically the intraorbital portion of the optic nerve shows some tortuosity and thickening (Fig. 9.3).
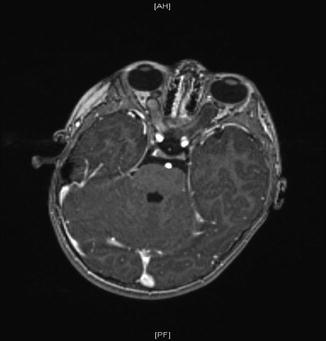
Fig. 9.3
Optic pathway glioma
Subependymal giant cell astrocytomas have the appearance of “snowballs” sitting in the ventricles (Fig. 9.4).
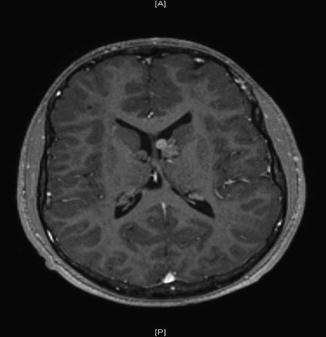
Fig. 9.4
Subependymal giant cell astrocytoma
Pathology
Low grade gliomas are grade I or II tumors as per the WHO grading system of astrocytic, oligodendroglial, and mixed glial–neuronal origin. They represent a heterogeneous group of tumors and as per the 2007 World Health Organization (WHO) classification of primary CNS tumors include the most common ones [3]:
(a)
Juvenile pilocytic astrocytoma (JPA), characterized through Rosenthal fibers (Fig. 9.5)
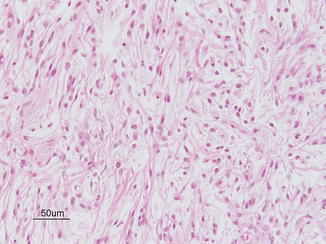
Fig. 9.5
Juvenile pilocytic astrocytoma. Courtesy of Cynthia E. Hawkins
(b)
Pilomyxoid astrocytoma (PMA), first named in 1999 by Tihan et al., describing a tumor typically located in the hypothalamic/chiasmatic region, affecting predominantly infants and younger children and appears to have a less favorable prognosis [4]. They are characterized histologically by a prominent myxoid matrix and angiocentric arrangement of monomorphous, bipolar tumor cells (Fig. 9.6).
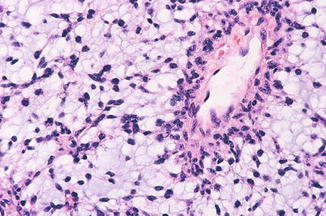
Fig. 9.6
Pilomyxoid astrocytoma (PMA). Reprinted from Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathologica, 2007; 114: 97–109. With permission from Springer Verlag
(c)
Diffuse (fibrillary, gemistocytic, protoplasmic) astrocytoma, this subtype is the most common in the adult population. Fibrillary astrocytomas stain positively for glial fibrillary acidic protein (GFAP). Some pediatric series describe fibrillary astrocytoma as the second most common subtype [1] (Fig. 9.7).
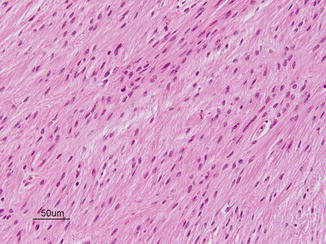
Fig. 9.7
Fibrillary astrocytomas. Courtesy of Cynthia E. Hawkins
(d) Pleomorphic xanthoastrocytomas (PXA) are typically superficial cortical tumors with a large cystic component arising from cortex. They are uncommon, but important to recognize as usually WHO grade II tumors. The neoplasm consists of sheets of large pleomorphic cells with abundant pink cytoplasm and focal perivascular collections of lymphocytes. Cytoplasmic vacuolization due to the presence of lipid accumulation is often present in the bizarre cells, although it is not a prominent feature of this case. Rosenthal fibers and eosinophilic granular bodies are sometimes found. Mitotic activity, microvascular proliferation, and necrosis are usually absent. They can often be cured with a gross total resection alone (Fig. 9.8).
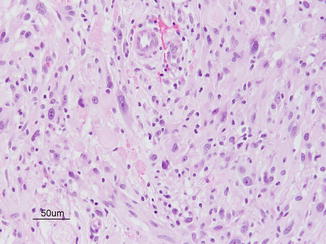
Fig. 9.8
Pleomorphic xanthoastrocytomas. Courtesy of Cynthia E. Hawkins
The WHO grading is very important for treatment and prognosis. As WHO grade II tumors tend to have a more aggressive behavior, the role of adjuvant treatment has to be considered carefully as the risk of further progression is higher.
Despite being the most frequent brain tumor in childhood, there are hardly any data on molecular biology available. Currently the main focus is on BRAF gene mutations [5, 6], which were initially described in 2005 and have been described in other malignancies as well like malignant melanoma. BRAF is a proto-oncogene that works as a downstream target for the RAS protein. The RAS (MAPK) signaling pathway plays a significant role in cell growth, differentiation, and apoptosis. So mutations or duplications of the BRAF gene will lead to oncogenic activation (Fig. 9.9).
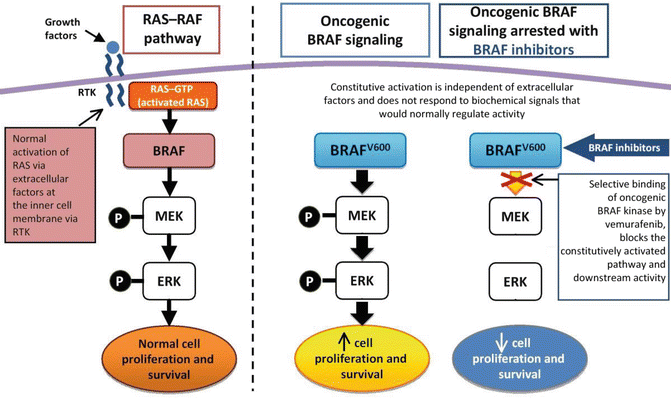
Fig. 9.9
BRAF gene. Reprinted from Ascierto PA, Kirkwood JM, Grob J-J, et al. The role of BRAF V600 mutation in melanoma. Journal of Translational Medicine 2012; 10(1):85. With permission from BioMed Central Ltd.
The majority of LGA (low grade astrocytoma) have the KIAA-1549 fusion gene as demonstrated below (Fig. 9.10).
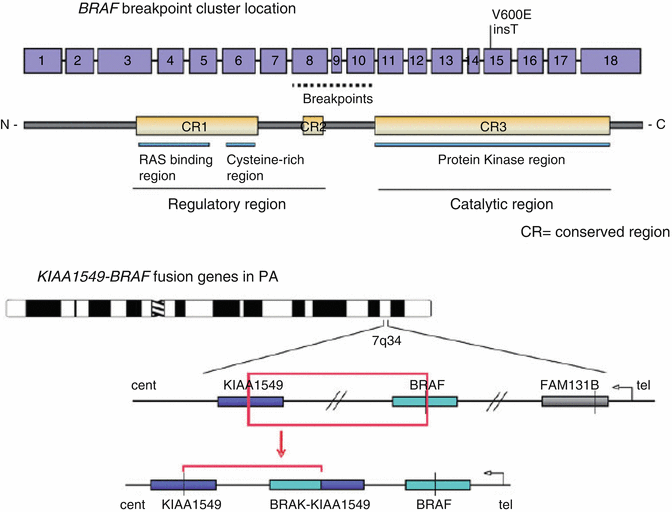
Fig. 9.10
Graphic breakpoint of the BRAF gene. Reprinted from Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 2011; 121(6): 763-774. With permission from Springer Verlag
The clinical relevance of this finding has been studied in a paper by Hawkins et al. [7]. They correlated tumor samples with BRAF alterations with clinical data retrospectively and the fusion of the BRAF-KIAA-1549 has been identified as a positive predictive marker in an incompletely resected LGA independent of their location, pathology, or age. This pathway could serve as a possible drug target for treatment.
Spontaneous growth arrest can be observed in LGA. It has been speculated that the lack of telomere maintenance is responsible for this biological behavior [7]. Telomeres are found at the terminal end of each chromosome. They are protective, but get shortened which each mitotic cycle until a growth arrest state. This study demonstrated that the telomeres of younger astrocytoma patients were significantly longer resulting in a more aggressive behavior and further recurrence. The conclusion was made that telomere maintenance could function as a biological prognostic marker.
Treatment
Surgery is the main treatment modality and curative when complete removal is possible with acceptable functional outcome [8]. This mainly applies to tumors within the cerebellum or hemispheric tumors.
Since surgery for deep-seated tumors is often restricted to debulking procedures or image-guided biopsies, focal radiation therapy was traditionally applied to such tumors. Over the years the lower age limit for radiation as a first-line adjuvant therapy has been changed to older children (over 10 years of age), but between different study groups no consensus has been reached in regard to the optimal age to continue deferral of radiation. This is likely dependent at least in part to the degree of dysfunction from the tumor and the potential radiation volume. Also more knowledge not only about neurocognitive impairment but also vascular complications, e.g., cavernomas, Moyamoya disease, or endocrinopathies, has emerged. Merchant et al. [9] published the largest prospective series on late effects for conformal radiation for low grade gliomas. Most of the tumors were localized in the diencephalon. Most of these patients needed multiple hormone replacements including growth hormone, thyroid, glucocorticoid, and gonadotropin-releasing hormone replacement. IQ decline was more severe with younger age at radiation and did not plateau 5 years post radiation treatment.
Merchant et al. described in a prospective series disease control with a 5-year event-free survival at 87.4 ± 4.4 %, but raised the concerns of higher rates of vasculopathy in the younger age group less than 5 years of age [10].
After initial anecdotal observations in the 1970s, chemotherapy has gained an important role as primary treatment of LGG, particularly in young children. Most of the early series were using a single-agent approach with either alkylating or platinum-based chemotherapies. More recently, combination therapies over extended periods were developed to potentially address the biological pattern of these slow growing tumors.
The initial indication for the use of chemotherapy was a salvage approach in the setting of a progressive tumor following radiation treatment. However, several prospective studies demonstrated safety and efficacy of chemotherapy as the first-line treatment for LGG. The primary goal with this approach is to avoid or at least postpone radiotherapy, which will then be reserved for recurrent/progressive tumors. Since radiation therapy is known to affect neurocognitive and endocrine function and growth and may trigger malignant transformation, there is increasing interest in considering repeated chemotherapy at further progression of LGG, particularly in young children.
The most commonly used combination (North America and Europe) is still carboplatin and vincristine (CV). Another well-established regimen is TPCV (thioguanine, procarbazine, lomustine, and vincristine), which has been tested in a randomized study versus CV [11]. The treatment adapts to the natural growth of the tumor, so it is low dose over a prolonged period of time (52 weeks). The goal of the treatment is to prevent further growth and stabilize the size of the tumor to avoid further damage. A 5-year event-free survival has been reported at 39 % ± 4 % for the CV group compared to 52 ± 5 % for the TPCV group in patients less than 10 years of age (p = 0.1). Response rate in both arms were similar with progressive disease in 32 %. The TPCV arm had slightly more toxicity when allergic reactions to carboplatin were excluded. The German HIT-LGG study group reports a 10-year event-free survival of 44 % with a slightly different CV regimen [12].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


