Fig. 12.1
CT and MR imaging of medulloblastoma.(a) Axial contrast-enhanced CT showing a hyperdense mass compressing the 4th ventricle (white arrow). Note the hydrocephalus with accompanying trans-ependymal flow (white arrow). (b) Axial T1 gadolinium-enhanced MRI and (c) axial FLAIR showing an enhancing mass in the midline cerebellum. (d) Axial T1 and (e) accompanying axial diffusion coefficient map showing restricted diffusion. (f) Sagittal T1 gadolinium-enhanced MRI through the midline spine showing laminar leptomeningeal enhancement along the dorsal and ventral aspects of the spinal cord and ventral brainstem indicative of metastatic dissemination (arrows)
Histopathology
Microscopically, MB is often referred to as a “small round blue cell tumor,” given the characteristics of densely packed cells with prominent nuclei surrounded by scant cytoplasm under H&E staining. The WHO classification of tumors of the central nervous system identifies five major histological variants in MB: classic, desmoplastic, large cell, anaplastic, and medulloblastoma with extensive nodularity (MBEN) [5]. Large cell and anaplastic variants have been correlated with poorer patient outcomes, while the best survival has been reported in patients with desmoplastic or MBEN histology; however, prognostic correlations may be influenced by age and other clinical features. Furthermore, as histologic categorization may be influenced by tumor heterogeneity, patient prognostication based on histopathological findings alone may not be accurate. The histologic differential diagnosis for MB includes atypical teratoid/rhabdoid tumor (ATRT) and rare embryonal tumors such as ETANTR [6]. Both entities which arise predominantly in younger children should be part of the differential work-up in all young patients with suspected MB. As characteristic rhabdoid cells may be present in variable amounts in ATRTs, immunostaining for the IN1/SNF gene product, which is nearly universally absent in ATRT but retained in MB tumors, should be included in the diagnostic work-up. Most but not all ETANTR exhibit classic histologic features of ependymoblastic rosettes and neuronal differentiation on a neuropil background and can be differentiated from MB by strong immuno-positivity for LIN28 and/or genomic amplification of the C19MC locus on 19q13.42 [7, 8].
Current MB Staging and Risk Stratification
Traditionally, MB patients are assigned into different treatment risk groups according to clinical features, which include age, the extent of resection, and the presence of metastasis at time of diagnosis. High-risk patients are defined as those <3 years of age, with more than 1.5 cm postsurgical residual tumor or evidence of metastasis at presentation [3]. Up to one third of MB patients present with leptomeningeal metastasis to the brain and/or spine, which may be detected on a preoperative MRI scan of the brain and spine. Postoperative MR imaging of the brain should be performed as soon as possible after surgery to avoid postoperative artifactual imaging changes to assess tumor residual. Staging is completed with cytological examination of the cerebrospinal fluid (CSF) for evidence of microscopic dissemination. Patients with nonmetastatic disease and no significant postoperative residual tumor, who are greater than 36 months of age, are stratified as standard risk.
Currently, all MB patients receive risk-adapted multimodality protocols based on clinical risk stratification schema. However, it is now increasingly clear that stratification based on clinical assessment alone is inadequate. It has been argued that current staging system fails to detect the true extent of disease and results in frequent over-/undertreatment of patients. Devastating acute and long-term treatment sequelae in survivors are major concerns for MB patients treated with intensive chemoradiotherapeutic treatment. Conversely, even with aggressive therapies, up to 30 % of MB patients will succumb to their disease. The lack of reliable clinical predictors of MB outcome has prompted substantial studies to identify biological predictors of clinical phenotype in MB.
Molecular Features of MB
MB was first linked to abnormalities in the Wingless (WNT) and Sonic Hedgehog (SHH) developmental signaling pathways based on observed association of MB with Turcot syndrome and Gorlin’s syndrome, and demonstration of alterations, respectively, in the APC and PTCH genes in some MB [9]. Early small cohort studies also suggested that specific genetic alterations, notably, MYCC gene amplification and CTNNB1 mutations, had prognostic correlations in MB (reviewed in ref [10]). Recent global gene expression and copy number studies of substantial cohorts of MB have helped to consolidate these early findings and establish a molecular classification system for MB that correlates with clinical phenotypes and patient outcomes. Specifically several global gene expression profiling studies of substantial MB cohorts have now demonstrated that MB is comprised of four molecular variants termed WNT, Sonic Hedgehog (SHH), and group 3 and group 4 subtypes which are associated with distinct developmental pathway signatures and/or cytogenetic abnormalities. The 4 MB subgroups also correlate with distinct tumor histology, patient demographics, and survival [10]. The WNT subgroup exhibits the best prognosis of any subgroup (greater than 95 % survival) and typically occurs in older children and exhibit classic histology. SHH MB represents an intermediate prognosis subgroup with overall survival ranging from 60 to 80 % and is predominantly seen in infants and young adults; MB with desmoplastic histology is almost exclusively restricted to this subgroup. Group 3 and 4 tumors which are not associated with any specific developmental signatures have the worst overall survival. MYCC gene amplification was observed only in group 3 tumors which also display frequently anaplastic histology, while group 4 tumors commonly (>30 %) exhibited isochromosome 17q (i17q, loss of chromosome 17p and gain of 17q). Characteristics of these subgroup variants are summarized in Fig. 12.2 [10]. These findings have helped to significantly advance our understanding of MB molecular biology and provided valuable diagnostic and prognostic tools that are currently in consideration for use in up-front risk stratification of patients in clinical trials across North America and Europe. A significant challenge is the development of robust diagnostic assays that can be used in clinical trials to reliably distinguish MB subgroups with high sensitivity and specificity. Currently, most consistent results to subtype MB have been reported with the use of nuclear CTNNB1 immuno-positivity and monosomy 6 which identifies WNT MB and MYCC amplifications which identifies group 3 MB; however, assays to reliably identify SHH and group 4 MB are lacking. Promising assays for MB subgrouping which include immunostains for SFRP1 in SHH, NPR3 in group 3, and KCNA1 in group 4 MB [11], and newer focused transcriptional and methylation assays remain to be validated.
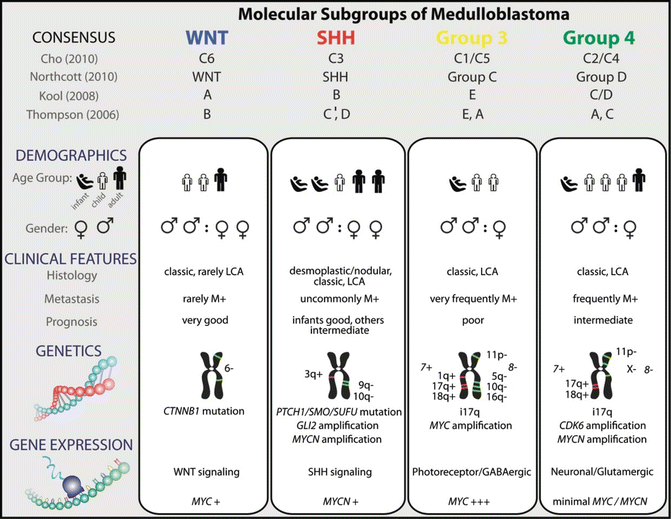
Fig. 12.2
Genetic, demographic, and clinicopathological features of the four molecular subgroups of medulloblastoma
The availability of tools to segregate molecular subtypes of MB will profoundly alter the design of MB clinical trials [11]. In addition to enhanced risk stratification for current conventional treatment regimens, molecular subtyping of MB will enable concerted investigations of novel therapy tailored to subgroup-specific biology. The inclusion of molecular analyses with traditional histo-clinical examination will be the standard of care in establishing the diagnosis and treatment stratification of MB in the near future.
Therapeutic Approaches
Multimodal approach of maximal safe surgical resection, radiotherapy to the primary tumor site and craniospinal axis, and systemic adjuvant chemotherapy are the current standard of care. Patients with MB often present with significant obstructive hydrocephalus, and thus management of increased intracranial pressure is commonly the priority. Patients may be managed with corticosteroids to alleviate tumor edema or require CSF diversion prior to surgery. With current treatment strategies, an anticipated 5-year overall survival (OS) has reached up to 80 % for patients with localized disease. However, metastatic and recurrent MBs still result in a significant mortality.
Surgery and Radiotherapy
Maximal safe resection of the posterior fossa mass is a key component and goal for patients with MB. With modern surgical techniques, gross total resection can be achieved in a majority of patients. As residual disease is associated with poorer outcome, immediate reoperation or second look surgery after adjuvant chemo- or radiotherapy may be considered.
Radiotherapy remains a critical part of the multimodal approach and is delivered early, within a month postsurgery, as delayed radiation results in poorer outcome [12]. The goal of radiotherapy is to control for both residual microscopic tumor in the primary site and to treat or prevent leptomeningeal disease along the craniospinal axis. Due to the severe toxic effects of irradiation to the developing nervous system, craniospinal radiation is often avoided or delayed in patients under the age of 3. Children with both average- and high-risk MB patients receive the same dose of local tumor bed irradiation of 5,400–5,580 cGy, but receive risk-adapted craniospinal irradiation. Children without tumor residual or metastasis received a 2,340 cGy craniospinal irradiation, while high-risk patients receive at least 3,600 cGy to the neuraxis [13]. Further reduction of craniospinal irradiation to 1,800 cGy for average-risk patients is currently being investigated in a phase III randomized control study by the North American Children’s Oncology Trial Group (COG).
In European and North American trials, the standard of care for MB involves postsurgical radiation followed typically by adjuvant cisplatin-based or high-dose chemotherapy. With such regimens, a 5-year survival for average-risk MB patients has reached 75–85 %; however, survival of high-risk patients is significantly poorer with a 5-year OS of 30–65 %. Significant improvement in survival has been achieved with chemoradiotherapy combination regimens; however, high-dose craniospinal radiotherapy continues to be associated with a high incidence of treatment-related complications. In addition to cognitive impairment, ototoxicity, thyroid dysfunction, growth failure, and endocrine abnormalities are significant sequelae in MB survivors. Intensity-modulated and proton-based radiation therapies represent promising newer normal tissue-sparing radiotherapeutic approaches. In addition to the use of conventional concomitant chemotherapy, development of novel radiosensitizers would be an important step toward minimizing radiation-associated toxicity.
Chemotherapy
Adjuvant chemotherapy plays an important role in the management of both average- and high-risk MB patients and has been used with the intent to permit reduction in radiation doses for older children or to avoid or delay radiation in younger children.
Radiation-sparing approaches for younger children with MB have been varied both in terms of chemotherapeutic regimens and inclusion of age range. In addition, some groups have used focal radiation up-front. Global trial groups have used three general approaches in children <3–5 years of age with MB to achieve a 5-year OS ranging between 50 and 70 %. These regimens have generally differed in the use of methotrexate, intraventricular treatment, and use of high-dose chemotherapy for consolidation. While the SFOP [14] group has used chemotherapy-based regimens without methotrexate, the UKCCSG/SIOP [15] and the German trial group protocol [13] employed a methotrexate-based chemotherapy backbone with intraventricular methotrexate treatment in the German experience. The Head Start consortium, which pioneered the use of high-dose chemotherapy, currently employs a methotrexate-based induction regimen with a single high-dose/stem cell rescue as consolidation [16], while treatment in the COG protocol is consolidated with 3 cycles of high-dose therapy and stem cell rescue [17]. As favorable outcomes with desmoplastic MB in young children have been seen across different infant MB studies, planned trials are examining desmoplasia as a criterion for stratification. An ongoing COG high-risk infant MB/CNS-PNET protocol is examining the benefits of methotrexate during induction in high-dose chemotherapy-based regimens, while the relative merits of single versus three stem cell rescue in consolidation remain to be investigated.
Treatment approaches to older children with MB have been more consistent. With the exception of a stem cell-based protocol from St. Jude’s, most trial groups have employed a conventional chemotherapy-based regimen with similar drugs. For average-risk patients, over the age of 3, both the COG and SIOP trial groups have employed a chemotherapy-based regimens with CCNU, VCR, and cisplatin, followed by reduced dose 2,340 cGy craniospinal irradiation and reported similar 5-year EFS, respectively, of 81 % [18] and 77 % [19]. In the St. Jude Medulloblastoma-96 protocol where standard-dose craniospinal in radiation is followed by four cycles of cyclophosphamide-based, dose-intensive chemotherapy, 5-year OS of 85 % has been reported [20], suggesting that dose intensification may benefit some average-risk patients.
Overall survival of patients with high-risk disease, specifically those presenting with metastasis, has been less favorable with 60–65 % long-term survival observed across chemotherapy and high-dose-based regimens. However, improved results have been reported by the Milan strategy in which patients received postoperative methotrexate, etoposide, cyclophosphamide, and carboplatin in a 2-month schedule, followed by hyperfractionated accelerated radiotherapy (HART). This regimen resulted in a 5-year OS of 73 %. COG also reported better survival in a phase I/II trial for high-risk MB in which patients received 15–30 doses of carboplatin along vincristine as radiosensitizers, followed by cisplatin-based maintenance chemotherapy. This study reported the highest overall survival at 82 % in high-risk MB patients who received 6 months of maintenance chemotherapy with cyclophosphamide thus suggesting a role for biologic-based maintenance chemotherapy in MB therapy [21].
Due to the increased incidence of secondary malignancies from irradiation and chemotherapy, periodic surveillance with brain and spine MRIs for disease recurrence, as well as secondary malignancies, is performed. In addition, regular neuropsychological and medical surveillance for end-organ toxicity is indicated for MB survivors. Recurrent disease, which occurs in approximately 25 % of patients with MB, remains a significant clinical challenge. Most relapses tend to occur within the first 3 years post-diagnosis, and long-term survival in this population remains very poor, with no clear effective rescue regimens reported. Limited successes have been reported with high-dose chemotherapy/autologous stem cell rescue regimens in older children with recurrent MB [22]. Higher salvage rates have been reported in younger children, who have not received prior irradiation, with radiation-based rescue protocols [23].
Molecular Therapeutic Targets
Basic research has led to tremendous gains in biological knowledge regarding MB. Specifically, the establishment of molecular subgroups for MB and the development of multiple group specific animal models are poised to transform future clinical trials and treatments for MB patients. It is expected that in the near future, patients will be stratified and treated based on the biological subgroup-specific makeup of their disease, which will hopefully lead to improved outcomes with less adverse effects. One main goal is to reduce morbidity of current treatment regimens in children with favorable biology disease. Specifically reducing chemotherapy and craniospinal irradiation for the favorable WNT subgroup could be one tangible approach that will minimize treatment toxicity.
To date, multiple pharmacological inhibitors for SHH-driven MB subgroups have been designed and have shown promising antitumor effects in SHH MB mouse models, and some are currently under clinical trials (Table 12.1). Promising preclinical studies, however, have not correlated with sustained disease response due to development of drug resistance with monotherapies [25]. These observations suggest that a combination of targeted therapies with conventional chemotherapy regimens may be required for optimal efficacy. Additionally, the use of several biologic agents that target signaling cross talk between SHH signaling and other key molecular pathways, such as AKT, Notch, TGF-β(beta), may also offer novel approaches to tailored therapy [31]. The feasibility of employing combination signaling therapies has been assessed in MB preclinical models. For instance, retinoic acid, which induces apoptosis together with histone deacetylase inhibitors, exhibits synergistic effects in xenograft and transgenic models [32]. Furthermore, a combination of LDE225 with PI3K inhibitors also markedly delays development of resistance thus suggesting the importance and promise of multiple pathway inhibition for sustained tumor response [26].
Table 12.1
Examples of preclinical pharmaceutical agents for targeted therapeutics in medulloblastoma
Drug name | Mode of action | Stage | Reference |
|---|---|---|---|
GDC-0449 | Smo inhibitor | Phase I | |
LDE225 | Smo inhibitor | Phase II | [26] |
Lapatinib | ERBB2 inhibitor | Phase II | [27] |
PHA665752 | MET inhibitor | Preclinical | [28] |
Tipifarnib | Farnesyltransferase inhibitor | Preclinical | [29] |
IPI-926 | Smo inhibitor | Preclinical | [30] |
Recent studies implicate the PI3K/AKT pathway, an effector and downstream target of MYCN/C, respectively, in SHH and group 3 MB biology. Thus, PI3K/AKT inhibitors also represent attractive new drug approaches for these patients [33]. Indeed small molecule inhibitors of PI3K/AKT have been demonstrated to suppress MB tumorigenesis in in vitro cell culture conditions [34], as well as in MYC-driven mouse models [33]. As MYC overexpression confers aggressive and metastatic behavior in MB [35], there has been considerable interest in directly targeting MYC or altering MYC downstream effects. Intriguingly, recent studies in myeloma and lymphoma models have shown the effective use of BET bromo-domain inhibitors to suppress MYC expression [36]. More interestingly, recent focus on screening synthetic lethal targets for MYC-driven cancer also identified potential new targets such as the core SUMOylation machinery and eukaryotic initiation factor complex assembly that are required to support MYC oncogenic state [36], for therapeutic opportunities. It is anticipated that with further dissection of MB subgroups and development of more precise subtype models that treatment of MB will approach a truly tailored approach with regimens that incorporate a spectrum of biologic agents that will improve patient outcomes with minimal sequelae.
Central Nervous System Primitive Neuroectodermal Tumor/Pineoblastoma
Epidemiology
Central nervous system primitive neuroectodermal tumors (CNS-PNETs) encompass a collection of embryonal tumors which are poorly differentiated with varying degrees of neuronal, astrocytic, or ependymal differentiation. They represent ~2.5 % of all childhood brain tumors [5] and are predominantly hemispheric in location but can also arise in multiple other CNS locations including the posterior fossa. In the most recent WHO classification system, CNS-PNETs are subcategorized based on location and histology and include CNS-PNET-NOS or supratentorial-PNET which are hemispheric tumors without any distinctive histologic features that overlap with tumors previously labeled as SPNET. Other categories identified by specific histologic features include CNS-neuroblastoma, CNS-ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma. Cerebral neuroblastomas and ganglioneuroblastoma, respectively, display only neuronal differentiation or with the presence of ganglion cells. Medulloepitheliomas are rare and diagnosed based on the presence of features resembling embryonic neural tube formation, while ependymoblastoma are characterized by distinct multilayered “ependymoblastic” rosettes; however, accuracy and specificity of this subclassification is under debate [5], and ependymoblastomas has been proposed to overlap with the recently described aggressive ETANTR/EMTR histologic entity [6]. These observations highlight the significant challenge in histological classification of CNS-PNET, particularly for subtypes which exhibit closely related variant histology. Emerging data suggest these may also represent closely related biological and molecular entities. CNS-PNETs diagnosis and classification have been challenging and remain in flux. Notably, pineal region PNET has been considered and treated as SPNET in clinical trials; however, their biologic relatedness to tumors currently classified under the CNS-PNET umbrella remains unclear. A full understanding of the molecular spectrum of this broad category of CNS-PNET and their relationship to tumors restricted to the pineal region is critical for development of more specific diagnostics and therapeutics.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


