Fig. 13.1
This MRI shows a very large posterior fossa ependymoma filling the fourth ventricle and extending into the left cerebellopontine angle through the foramen of Luschka (Reprinted from Teo C, Nakaji P, Symons P, et al. Ependymoma. Child’s Nervous System 2003; 19(5–6): 270–285. With permission from Springer Verlag)
Supratentorial tumours tend to be more heterogeneous, with cystic, calcified and haemorrhagic components. They usually have avidly enhancing areas interspersed with nonenhancing regions (Figs. 13.2 and 13.3).
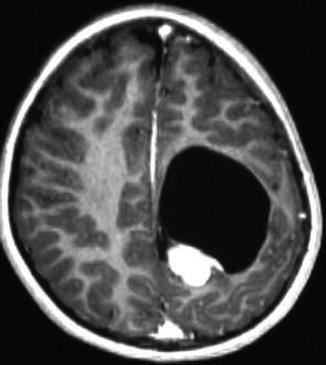
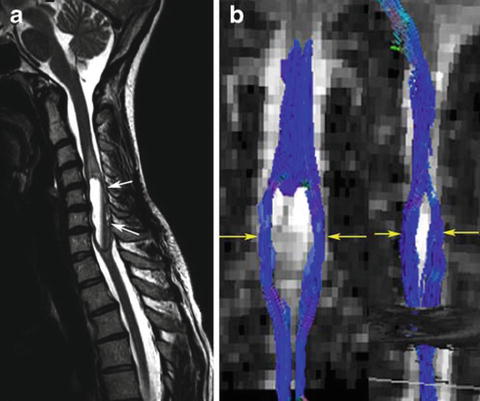
Fig. 13.2
Supratentorial ependymoma. The location makes these tumours more readily resectable. This patient did not have adjuvant therapy and has enjoyed 4 years of recurrence-free survival (Reprinted from Teo C, Nakaji P, Symons P, et al. Ependymoma. Child’s Nervous System 2003; 19(5–6): 270–285. With permission from Springer Verlag)
Fig. 13.3
Ependymoma: there is a tumour in the lower cervical spinal cord (a) with displacement of the fibres visible on the tractogram (b) (Reprinted from Vargas MI, Delavelle J, Jlassi H, et al. Clinical applications of diffusion tensor tractography of the spinal cord. Neuroradiology 2008; 50(1): 25–29. With permission from Springer Verlag)
Pathology
Histological classification of ependymomas is most frequently defined using the World Health Organization (WHO) grading scheme for CNS tumours [2]. According to this system, ependymomas can be divided into four pathological subgroups: subependymoma and myxopapillary ependymoma (both WHO grade I), classic (grade II) and anaplastic (grade III). Within the classic subgroup are four morphological variants: cellular, papillary, clear cell and tanycytic tumours (Table 13.1).
Table 13.1
Current WHO classifications of ependymomas
WHO grading system | Subgroups | Histopathology |
|---|---|---|
I | Subependymoma | Isomorphic nuclei embedded in a dense fibrillary matrix of glial cell processes with frequent microcystic change |
Myxopapillary ependymoma | Mitoses rare or absent | |
GFAP-expressing cuboidal to elongated tumour cells arranged in a papillary manner around vascular stromal cores | ||
II (classic) | Cellular ependymoma | Monomorphic nuclear morphology |
Papillary ependymoma | Mitoses rare or absent | |
Clear cell ependymoma | Perivascular pseudorosettes and ependymal rosettes | |
Tanycytic ependymoma | ||
III (anaplastic) | High mitotic activity. Palisading necrosis. Microvascular proliferation. Perivascular pseudorosettes |
Subependymomas are benign nodules usually found in the intracranial ventricles of adults. Similarly, myxopapillary ependymomas are rarely seen in children and tend to arise almost exclusively in the spine, at the level of the cauda equina. Paediatric intracranial ependymomas are typically of a classic or anaplastic histology. Hallmark microscopic features of classic ependymomas include either perivascular pseudorosettes, formed by the radial arrangement of tumour cells around a blood vessel (perivascular), or true rosettes where the cells circumferentially surround a central lumen or canal [3]. Positive cytoplasmic staining for glial fibrillary acid protein (GFAP) and vimentin is a frequent immunohistochemical finding. Anaplastic ependymomas are additionally differentiated by displaying numerous mitotic figures, substantial necrosis, an increased cellular nucleus/cytoplasmic ratio and microvascular proliferation. Assigning a specific WHO grade to a given tumour remains subjective, lacking uniformity, and is often difficult as ependymomas frequently demonstrate marked intratumoral heterogeneity. This is emphasised by the reported incidence of anaplastic ependymoma, varying from 7 to 89 % of cohorts analysed.
Electron microscopy may be used to confirm the histological identification of ependymoma; characteristic features include cilia, microvilli, long junctional complexes, basal bodies and intracytoplasmic filaments [4].
Biology
Recent times have witnessed an increase in studies aiming to improve our understanding of the nature and origins of molecular genetic abnormalities found in ependymoma. What is becoming apparent is that ependymoma is a biologically diverse and complex entity which, akin to medulloblastoma, appears to be comprised of biologically and functionally distinct molecular subclasses.
Predisposition Syndromes
The majority of paediatric ependymomas arise spontaneously without a known predisposition syndrome. Nevertheless, associations with certain conditions have been reported. For instance, neurofibromatosis type 2 is an autosomal dominant condition caused by a germline mutation in the NF2/Merlin gene (22q12.2). This disorder is particularly associated with the formation of vestibular schwannomas, although spinal ependymomas can be a relatively common finding [5]. Intracranial ependymomas have been described as a rare feature of Turcot syndrome type 2 [6]. This familial disorder is caused by inherited mutations of the APC gene (5q22.2) and is characterised by the development of intestinal polyps, yet is more commonly associated with medulloblastoma formation. Familial ependymomas occurring in the absence of such inherited syndromes have also been described, although these are extremely rare.
Cell of Origin
The morphological similarity between neoplastic ependymoma cells and normal ependymal cells lining the CNS ventricular system made the latter a presumed cell of origin for this tumour. However, recent stem cell characterisation studies have suggested that tumour initiation may actually arise in primitive radial glial cells (RGCs), which demonstrate both stem (CD133+/Nestin+) and glial (Rc2+/Blbp+) immunophenotypes. These RGCs have been shown in vitro to self-renew, and differentiate, whilst also forming ependymomas when transplanted into immunocompromised mice, making them attractive candidates [7, 8]. If this concept proves correct, it may lead to a shift in therapy by targeting these ependymoma-initiating cells in addition to reducing tumour bulk.
Molecular Subgroups
Earlier biological studies have shown distinct molecular differences between spinal, supratentorial and posterior fossa ependymomas, suggesting tumour location may be an important driving factor for these lesions [7]. Whilst the precise number remains uncertain, up to nine molecular subclasses of ependymoma from these three sites have now been suggested according to a combination of intrinsic chromosomal abnormalities and characteristic ‘driving’ gene mutations (such as NOTCH1 or EPHB2 amplification). For instance, at least two molecularly distinct subclasses of posterior fossa ependymoma have been confirmed, each associated with a specific patient age group and clinical outcome [9]. Likewise, supratentorial tumours can be divided into two groups according the expression of genes controlling neuronal differentiation [10].
There is also evidence from chromosome imbalance work that childhood and adult ependymomas are biologically distinct, irrespective of tumour location, with gain of chromosomes 1q, 7 and 9 and loss of chromosomes 22, 3 and 9p being frequent in paediatric tumours [11]. Moreover, a ‘balanced’ genomic profile, without chromosomal gain or loss, has been reported in up to 58 % of paediatric ependymomas, particularly those under 3 years of age. This contrasts with adult ependymomas where a balanced genome is found in less than 10 % of cases [11].
Prognostic Factors
Due to the relatively poor survival outcomes of ependymoma when compared with other paediatric CNS tumours, there remains great interest in determining prognostic factors for this tumour group in order to risk stratify patients and tailor therapy appropriately. Several clinical, pathological and biological correlates of outcome have been assessed, primarily using data from retrospective cohort studies.
Clinicopathological Factors
Surgical Resection
The degree of tumour removal, particularly a gross total resection (GTR), appears the most favourable and consistent clinical prognostic factor reported from retrospective case series, with 5-year survival rates being reported as high as 80 %, compared to 22–47 % for cases of incomplete resection [11–13]. However, literature suggests that for approximately one-third of childhood intracranial ependymomas, GTR is not achieved. This is particularly the case for infratentorial tumours extending to involve the lower cranial nerves and associated vasculature, where aggressive resection can be associated with substantial morbidity.
Tumour Location
There is insufficient evidence that intracranial location impacts on patient prognosis, independent of that explained by surgical accessibility and the ease of achieving a GTR. There is data, albeit limited, to suggest complete resection for certain supratentorial ependymomas can be curative [14]. Indeed, the North American Children’s Oncology Group (COG) currently recommends only a post-operative observation strategy for grade II supratentorial tumours where GTR has been achieved.
Age
Historically, a younger age at diagnosis has frequently been associated with shorter patient survival. Possible explanations for this include an age-related difference in underlying tumour biology, or the practice of delaying or avoiding adjuvant radiotherapy to the developing central nervous system. However, whilst the majority of studies reveal an association with age, this is not consistent as some studies report no prognostic difference between contrasting paediatric age groups [15].
WHO Grade
A distinct difference in survival outcomes between anaplastic and classical ependymomas, according to the WHO grading system, remains unproven. This probably reflects the difficulty of applying such a classification scheme in clinical practice due to intratumoural heterogeneity, inter-observer variability and the inability to uniformly define anaplasia. Whilst some studies have observed an adverse prognosis for anaplastic ependymomas, other studies have refuted this. Unfortunately, alternative classification schemes have also failed to prove either reproducible or prognostic [16].
Biological Factors
Molecular Subgroups
As stated, advances in tumour molecular analysis (high-throughput gene expression and genomic arrays, tissue microarray analysis by immunohistochemistry and fluorescence in situ hybridisation, etc.) have enabled new biological subgroups of ependymoma to be identified. For instance, gene expression profiling of posterior fossa ependymomas has delineated at least two distinct prognostic groups [9, 17]. One such set of tumours (termed ‘Group A’ or ‘1’) were often located laterally in the cerebellopontine angle, frequently demonstrated overexpression of genes including laminin-alpha 2 (LAMA2), or genes involved in mesenchymal/extracellular matrix development such as Tenascin-C (TNC), and were associated with a younger patient age, recurrence and metastasis. The contrasting group of ependymomas (‘Group B’ or ‘2’) were primarily located centrally, found predominantly in adolescents or adults and were less invasive. Consequently, the progression-free survival (PFS) and overall survival (OS) rates for Group A/1 ependymomas were significantly poorer than for Group 2/B tumours. Likewise, biological prognostic subgroups of supratentorial ependymomas have been identified according to the expression of genes implicated in neuronal differentiation such as NFL70, where overexpression independently conferred an improved PFS [10].
Chromosomal Imbalances
Gain of chromosome 1q is the most common chromosomal imbalance observed in paediatric ependymomas, when compared to adult tumours [11]. Nevertheless, it is only observed in approximately 20 % of cases [18]. Several retrospective genomic studies performed in both North America and Europe, including analyses of uniformly treated clinical trial cohorts, have confirmed an adverse prognostic role for 1q gain, being associated with a worse patient PFS and OS. Studies have also proposed incorporating 1q gain into future patient risk group stratifications in order to tailor therapy more appropriately [19]. The precise mechanism of 1q gain and why, in turn, this seems to impart a worse outcome for patients remains unclear.
Gain of another chromosomal region, 9q33-34, has been shown to occur relatively frequently at recurrence, in addition to being associated with an adverse prognosis in paediatric ependymoma. This region encompasses the loci of genes thought to be implicated in ependymoma pathogenesis such as NOTCH1 and TNC [18].
Individual Molecular Markers
Several biological markers have been proposed as correlates of adverse outcome in paediatric ependymoma from retrospective case series of paediatric ependymoma. These include overexpression of human telomerase reverse transcriptase (hTERT) [20] and its nuclear chaperone Nucleolin [21], the extracellular matrix proteins TNC and LAMA2 [9, 22], p53 [23], EGFR [24, 25], the transcription factor EVI-1 [26] and the stem cell marker Nestin [27].
However, all of these putative prognostic candidates have not been completely validated, either by generating reproducible findings in different retrospective cohorts or indeed in a prospective clinical trial setting. Until this is undertaken, definitive conclusions regarding their effectiveness as prognostic markers in paediatric ependymoma should be considered with caution.
Evidence has shown, however, that ependymoma can no longer be considered a single biological entity. Indeed, as suggested by Grill and colleagues, due to the biological complexity and diversity of paediatric ependymoma, future prognostic predictions may require a set of several biomarkers which have relevance for a particular molecular subset of the tumour, as is currently the case for medulloblastomas [18].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


