Fig. 23.1
Axial (a) and sagittal (b) brain MRI T1-weighted images with contrast showing a characteristic SMART syndrome. This 13-year-old female was treated with 39.6 Gy of craniospinal radiation following a diagnosis of medulloblastoma when she was 6 years of age. Seven years later she presented with headache, fever, aphasia, and right hemianopsia. CSF analyses were negative for herpes virus. She recovered completely but presented a total of four similar episodes
Secondary Neoplasms
While the overall survival in childhood cancer survivors increases, secondary neoplasms have emerged as a long-term complication of treatment. Two large registries and two population-based studies have estimated a cumulative incidence of subsequent malignant neoplasms (SMNs) in childhood cancer survivors of 3 to 4 % at 20 years posttreatment [16, 17]. Survivors of Hodgkin disease are afflicted with higher rates of breast and thyroid cancers, whereas leukemia and primary CNS tumors have tendency to develop a subsequent CNS tumor. Armstrong et al. reported 76 cases of SMNs in 1877 survivors of CNS malignancies with second CNS tumors (26 %), followed by thyroid (16 %) and sarcoma (11 %). They also observed 171 neoplasms classified as “benign” tumors including 59 meningiomas and 112 nonmelanoma skin cancers. The overall cumulative incidence of a subsequent neoplasm at 25 years was estimated to be 10.7 % (Fig. 23.2) [18]. Most common malignancies are malignant astrocytomas which are overrepresented by glioblastomas, followed by sarcomas and occasionally supratentorial primitive neuroectodermal tumors (sPNET) (Fig. 23.3).
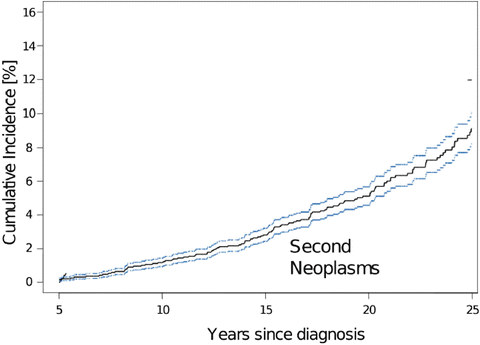
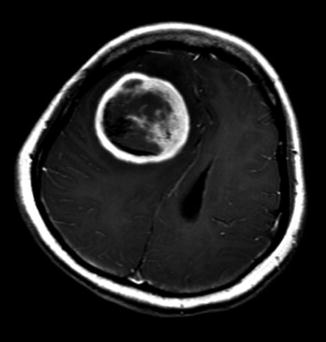
Fig. 23.2
Cumulative incidence of subsequent neoplasms in long-term survivors of childhood central nervous system tumor based on Childhood Cancer Survivors Study. [Reprinted from Armstrong, G.T., Long-term survivors of childhood central nervous system malignancies: the experience of the Childhood Cancer Survivor Study. European journal of paediatric neurology: EJPN: official journal of the European Paediatric Neurology Society, 2010. 14(4): p. 298–303. With permission from Elsevier]
Fig. 23.3
Axial brain MRI T1-weighted image with contrast showing a right frontal subsequent supratentorial primitive neuroectodermal tumor (sPNET). This 17-year-old female patient was treated 7 years earlier with craniospinal radiation for CNS relapse of acute lymphoblastic leukemia. Despite two surgeries, salvage radiation therapy and chemotherapy, she rapidly progressed and died 5 months after diagnosis
In a study based on an acute lymphoblastic leukemia (ALL) population, the mean time between diagnosis and development of subsequent CNS neoplasm was 11.9 years, with 20.6 years for meningiomas and 8.8 years for all other CNS tumors. In fact, the cumulative incidence of CNS SMNs such as glioblastomas plateaued at 15 years, whereas the cumulative incidence of meningiomas continue to increase beyond 35 years posttreatment [19].
Despite similar histological appearance, secondary malignancies typically behave more aggressively and are resistant to treatment. The overall survival for second glioblastomas is uniformly poor, and second sPNET have an overall survival of 18 % at 18 months compared to 40–50 % at 36 months for a primary sPNET [20]. Subsequent meningiomas have also been found to be more often atypical and prone to relapse.
The development of SMNs is most likely multifactorial, but RT certainly contributes to this process. The vast majority of SMNs develops within the radiation field. At 25 years, the cumulative incidence of SMNs was 7.1 % for patients who received more than 50 Gy to the cranium compared to 1 % for those who did not received RT. A linear dose response is observed with an excess relative risk per Gy of 0.33 for gliomas and 1.06 for meningiomas.
The exact contribution of chemotherapy to the development of SMNs is more difficult to assess partly due to the use of combination chemotherapy regimens. Alkylating drugs, especially cyclophosphamide and epipodophyllotoxins, such as etoposide, have been reported to increase the cumulative incidence of second malignancies up to 4 % compared to 1 % [21]. The majority were acute non-lymphomatous leukemia (ANLL) and acute myeloid leukemia (AML) which tended to occur within the first 3 years after treatment.
Somatic mutations can predispose an individual to develop SMNs. Patients with p53 (Li-Fraumeni syndrome) are more likely to develop sarcoma, primary brain tumor, and ALL after treatment. Therapy may be tailored in order to reduce exposure RT and certain chemotherapy agents, but usually those patients are diagnosed at their subsequent neoplasm. No study has tested systematically all patients with SMNs, but the prevalence of such mutation remains probably low. In six patients with ALL that eventually developed a second CNS tumor, none were carrier of a p53 mutation. Other somatic mutations such as the ataxia telangiectasia mutated gene (ATM) known to be involved in DNA repair possibly play a role. Whereas homozygote patients present a classical phenotype with childhood ataxia, telangiectasia, and immunodeficiency, heterozygotes are at increased risk (odds ratio 5.8) of neoplasm following exposure to radiation. Patients with NF1 are also more likely to develop a secondary malignancy. Sharif et al. reported that 20 % of patients with optic pathway glioma developed SMNs, and half of patients treated with radiotherapy developed an SMN, usually gliomas or malignant nerve sheet tumors [22]. Finally, metabolism and detoxification might also be involved in the development of SMNs especially in those with ANLL and AML. Polymorphisms of NAD (P)H:quinone oxidoreductase (NQO1), glutathione S-transferase (GST), and thiopurine S-methyltransferase (TPMT) have been reported. An ongoing cooperative group study from the Children’s Oncology Group aims to address those predisposing factors by enrolling patients with SMNs and assessing their p53, ATM, and polymorphisms status.
The role and benefice of surveillance brain MRI to detect asymptomatic SMNs remains unknown. Long–Term Follow–Up Program Resource Guide published in 2008 by the Children’s Oncology Group recommends conducting imaging only if the patient presents new-onset symptoms [23]. Experts however suggested surveillance brain MRI every 2 years for patient with NF1 who received RT.
Long-Term Neurological and Neurosensory Sequelae
Long-term survivors of childhood CNS tumors are at high risk of neurologic and neurosensory impairment not only during treatment but also years after completion of their therapy.
Seizures and Epilepsy
Epilepsy may represent a significant problem in a subset of young adult survivors. Studies have reported that between 1 % and 3 % of children with new-onset seizures have a brain tumor and that seizure is the initial presenting symptom at diagnosis in up to 9 % of children with brain tumor [24]. Patients with slowly growing neoplasms such as dysembryoplastic neuroepithelial tumor (DNET), ganglioglioma, pleomorphic xanthoastrocytoma, and cortically based low-grade astrocytoma are particularly prone to develop epilepsy. The vast majority (81 %) has a supratentorial tumor that generally involves the temporal lobe [25, 26]. Risk factors for poor outcome include having seizures for more than 1 year preoperatively and partial resection. Seizure freedom can be expected in 79 % of patients undergoing a gross total tumor resection versus 43 % for a partial resection. It is still unclearif there is a benefit to perform expanded-field epilepsy surgery with intraoperative electrocorticography in contrast to a brain tumor surgery, where the goal is to remove the tumor and obtain diagnostic tissue. In some instances, chemotherapy and RT have also been reported to improve seizure control.
In a report from the Childhood Cancer Survivor Study (CCSS), the prevalence of epilepsy in long-term survivors of childhood brain tumors was 25 %. Interestingly, 6.5 % had their first seizure more than 5 years after diagnosis of their cancer [27]. Seizures were more frequent in patients treated with RT > 30 Gy to any cortical area (relative risk [RR] 2) and more frequent in children treated at young age. Methotrexate has also been related to late seizure onset, especially in the setting of necrotizing leukoencephalopathy.
Seizure control with antiepileptic drugs appears similar to what can be expected in patients without brain tumor. In a report from Khan et al., 65 % were controlled and 17 % were considered intractable. Uncontrolled seizures were more frequent in children with neurologic deficits, pericavity T2 abnormalities on MRI, and slow waves on EEG [26]. However, another group reported that only 39 % were seizure-free and that 26 % were intractable [25]. For patients who are seizure-free with medication, a slow tapering can be attempted after 2–3 years. In one study, including 62 patients with pediatric brain tumors and epilepsy, 73 % who were weaned off remained seizure-free [26]. Unfavorable risk factors included multiple tumor resections and whole-brain RT.
Motor Dysfunction
Motor impairment is a well-known late-onset sequela, but few studies have specifically been conducted to address this subject. Multiple contributing factors include the tumor itself, hydrocephalus, surgery-associated morbidity, side effects of chemotherapy, and RT. Not surprisingly, it has been reported that compared to their siblings, 49 % of childhood brain tumor survivors had coordination problems and 26 % presented motor difficulties. The onset of those dysfunctions occurred mainly at diagnosis or during treatment. Nonetheless, 4.6 % reported that their impairments appeared more than 5 years posttreatment. Patients who had received at least 50 Gy were found to be at higher risk for motor problems (RR 2.0) compared to those who received less than 30 Gy [27]. Late-onset leukoencephalopathy and cerebral vasculopathy associated with radiation have been hypothesized as possible mechanisms for late-onset motor and coordination impairments.
Primary spinal cord tumors are rare in children and account for 4–8 % of all tumors from the central nervous system. However, due to their location, a significant proportion of patients have long-term sequelae such as weakness, dysesthesia, and sphincter dysfunction. In general, postoperatively 35–70 % will remain stable, 40 to 60 % will improve, and 5 to 40 % will worsen neurologically [28]. Around 40 % of those who deteriorated following surgery will improve within a month, but improvement can be expected up to a year [28]. Postoperative functional outcome is strongly correlated to preoperative neurologic state. The presence of an identifiable tumor plane at surgery, cystic tumor, and improvement in neurological symptoms before discharge has been reported to be associated with overall neurological improvement.
Although initially considered reversible, up to 50 % of patients with postoperative posterior fossa syndrome were found to have permanent neurological dysfunction including cranial nerve neuropathy, hypotonia, and ataxia.
Vincristine and cisplatin commonly used in treatment of patients with brain tumors are known for their neurotoxicity including peripheral neuropathy. Whereas most patients significantly improve following completion of their therapy, young adults continue to present evidence of neuropathy several years later. Long-term follow-up studies have shown that reduction in deep tendon reflexes, raised vibration perception threshold, and abnormal nerve conduction were found in 44 %, 55 %, and 35 %, respectively [29]. Despite the fact that symptoms have been reported to persist in 20–75 % of patients, most had no impact or limitation in their daily activities. The outcome of such peripheral neuropathy 20 to 30 years after treatment is however unknown. As this population is at risk of metabolic syndrome, diabetes-associated neuropathy could add significant morbidity.
Although not extensively studied, pain can also affect daily functioning of long-term survivors of childhood cancer. More than 10 years post diagnosis between 10 and 30 % of patients reported pain or abnormal sensation. 16.7 % were using prescription analgesic drugs and 21 % attributed their pain to cancer and its treatment [30].
Ototoxicity
In targeting the outer hair cells in the organ of Corti and the vascularized epithelium in the lateral wall of the cochlea, high doses of platinum have been reported to cause irreversible early- or delayed-onset hearing loss. These late complications are not only creating functional limitation but also affect speech development, learning, communication, school performance, social interaction, and overall quality of life. Ototoxicity is one of the most common platinum-related neurotoxic effects; it is characterized by a dose-dependent high-frequency sensorineural hearing loss with tinnitus. The variable reported incidence of hearing loss is related to the specific criteria used to define hearing loss, the younger age at start of treatment, the high cumulative doses of platinum compounds (>400 mg/m2 for cisplatin and carboplatin), and the use of concomitant ototoxic treatments including CNS radiation therapy [31, 32]. Ototoxicity has a general dose dependence, but with considerable interindividual variability. Genetic polymorphisms in enzymes responsible for platinum metabolism (e.g., glutathione S-transferase, thiopurine methyltransferase, catechol O-methyltransferase) may contribute to the substantial interpatient variability in the severity of hearing loss [33, 34]. In addition, higher levels of residual platinum, which can persist for years following treatment, may also contribute to the severity of ototoxicity. Ototoxicity after platinum chemotherapy can present or worsen years after completion of therapy. Radiotherapy (RT) to the normal cochlea or cranial nerve VIII can also cause sensorineural hearing loss. Cranial RT, when used as a single modality, results in ototoxicity when cochlear dosage exceeds 32 Gy. Young age, presence of a brain tumor, and/or hydrocephalus can increase susceptibility to hearing loss. The onset of radiation-associated hearing loss may be gradual, manifesting months to years after exposure. When used concomitantly with platinum, RT can substantially exacerbate the hearing loss associated with chemotherapy. RT to temporal lobe (>30 Gy) and to posterior fossa (≥50 Gy but also ≥30–49.9 Gy) was associated with an increased risk of problems with hearing sounds, tinnitus, and hearing loss requiring an aid [35]. Concurrent administration of platinum and RT results in synergistic ototoxicity, especially in the high-frequency speech range. Care should be taken to distinguish sensorineural hearing loss versus conductive hearing loss due to serous otitis media as seen after RT, which occasionally requires myringotomy. Early detection of ototoxicity in children may minimize the risk for severe impairment in the frequencies required for speech recognition. In children being treated with either cisplatin or carboplatin, extended high-frequency audiometry (i.e., at frequencies above 8 kHz) is more sensitive to early hearing loss than conventional audiometry. This procedure is readily carried out in children more than 5 years old. For younger children, distortion product otoacoustic emissions (DPOAEs) are also more sensitive than conventional audiometry. Various strategies have been considered to minimize platinum ototoxicity. The data were insufficient to support the routine use of otoprotective agents such as amifostine to prevent platinum ototoxicity. A major challenge to the development of otoprotective agents is the lack of consensus on the best ototoxicity assessment criteria, which should include QOL component. Radiation reduction dose to the cochlea has been investigated, including the use of three-dimensional (conformal) RT, intensity-modulated RT (IMRT), and more recently proton therapy [36]. Once treatment is completed, long-term audiometric monitoring should continue according to one of recognized pediatric late effects monitoring guidelines for appropriate management.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


