Leukoencephalopathies
Mark P. Gorman
Leukoencephalopathies comprise a clinical and radiographic heterogeneous group of disorders. All these disorders share the common features of neurologic dysfunction and preferential involvement of CNS white matter. Although white matter can be affected by many different processes, the term leukodystrophy is generally reserved for those with an identified or presumed genetic basis that is associated with a loss of previously formed myelin. Acquired causes of white matter dysfunction include infectious (such as encephalitis), inflammatory (such as acute disseminated encephalomyelitis and multiple sclerosis; see Chapter 556, nutritional (such as vitamin B12 deficiency), and neoplastic (such as astrocytoma) etiologies. When evaluating a patient with a suspected leukoencephalopathy, these acquired disorders should be considered and excluded with specific testing when clinically indicated.
MRI can help to distinguish genetic from acquired white matter disorders. Normal myelination starts prenatally and continues for decades, generally proceeding in a caudalto-rostral and central-to-peripheral pattern.1 Depending on a patient’s age, specific areas are expected to be myelinated, and others are unmyelinated. With myelination, the brain MRI appearance changes, with increasing T1 signal and decreasing T2 signal in the myelinated areas. In general, brain MRI in acquired white matter disorders shows asymmetric disturbances in this process, whereas the leukodys-trophies produce a symmetric pattern of abnormalities (see Fig. 556-2).
In the past, treatment of genetic leukoencephalopathies was largely restricted to supportive measures. However, hematopoietic stem cell transplantation (HSCT) has been increasingly used for some of these leukoencephalopathies. For most of these disorders, replacement of the deficient enzyme from donor cells, especially monocytes that can cross the blood-brain barrier, serves as the underlying therapeutic mechanism. This process is slow and inefficient, typically leading to an approximate 6- to 12-month period before CNS symptoms stabilize, and does not typically reverse existing deficits.2 Therefore, although useful in certain disorders, HSCT has also been disappointingly ineffective for patients with advanced symptoms and/or one of the more rapidly progressing leukoencephalopathies. In addition, HSCT is associated with significant potential morbidity and mortality. Thus, as the genetic and molecular bases for the leukoencephalopathy become increasingly delineated, it is hoped that more specific treatments, such as enzyme replacement therapy or gene therapy, will become available. At the time of the writing of this chapter, such therapies have been implemented on a small scale for few of the disorders.
There is a very long list of leukoencephalopathies, which can be broadly divided into disorders in which there is a permanent deficit in myelin deposition (hypomyelinating), disturbances of myelin formation (dysmyelinating), or loss of existing white matter (demyelinating).1 Although useful for defining disease mechanisms, these categories do not readily translate clinically. Therefore, this chapter will use a clinical approach, dividing the leukoencephalopathies primarily based on age of onset, associated symptoms, and MRI appearance (Figs. 576-1, 576-2). Given the frequent heterogeneity within some of the leukoencephalopathies and the ever-expanding list of disorders, this algorithm is an oversimplification, but can nonetheless be a clinically useful initial guide to testing.
With the list narrowed, clinicians can then use the MRI appearance to support or refute their clinical impression, and biochemical and genetic testing to confirm a specific diagnosis. With this approach, the clinician can avoid testing for every leukodystrophy, a costly endeavor that is becoming rapidly impractical due to the growing number of identified leukodystrophies. This chapter will focus on the more common and clinically important leukodystrophies, as well as those whose genetic basis has been recently identified. For an exhaustive review of all white matter disorders, with an emphasis on the radiographic findings, the reader is referred elsewhere.1
X-LINKED ADRENOLEUKODYSTROPHY
X-linked adrenoleukodystrophy (ALD) is an X-linked recessive disorder that affects approximately 1 in 21,000 males and has an overall frequency (male hemizygotes plus female heterozygotes) of approximately 1 in 16,800 in the United States.3 It affects all racial and ethnic groups.
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
Mutations in the gene ABCD1 on Xq28 cause ALD. ABCD1 encodes ALD protein, a peroxisomal transmembrane protein that is a member of the ATP-binding cassette transporter family. Most mutations are missense (61%), frameshift (23%), or nonsense (10%) mutations, with less than 10% due to deletions or insertions.4 Ninety-three percent of mutations are inherited, with only 7% forming de novo.4 As of May 2008, nearly 500 unique mutations had been reported to the X-Linked Adrenoleukodystrophy Database (www.x-ald.nl).
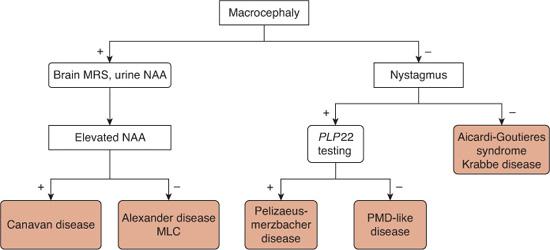
FIGURE 576-1. Algorithm for the differential diagnosis of leukodystrophies with onset in the first year of life. MRS, magnetic resonance spectroscopy; NAA, N-acetyl aspartate; MLC, megalencephalic leukoencephalopathy with subcortical cysts; PLP22, proteolipid protein 22; PMD, Pelizaeus-Merzbacher disease.
Although its precise activity is unknown, Adrenoleukodystrophy protein (ALDP) plays an essential, although indirect, role in the β-oxidation of very-long-chain fatty acids (VLCFA).5 Its dysfunction in patients with ALD leads to the accumulation of VLCFA in most tissues, particularly the CNS, adrenal cortex, and testes.
The presence of VLCFA in biologic membranes appears to alter their structure and function and may particularly impair myelin stability.6 White matter lesions in patients with ALD contain 3 zones: an outer layer with destruction of myelin but no inflammation, a central layer with demyelination and perivascular inflammation, and an inner layer with gliosis and little inflammation.5 Thus immune responses, particularly CD8+ cytotoxic T lymphocytes targeting oligodendrocytes, are important in ALD pathogenesis.7 Although the levels of VLCFA and the degree of inflammation appears to correlate in postmortem brain tissues of patients with ALD, the mechanisms linking these 2 processes are undefined.6
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Although many different phenotypes have been reported, most male patients with ALD present with the childhood cerebral (35% of all affected individuals), adrenomyeloneuropathy (40–45%), or “Addison disease only” (10%) forms.4 The cerebral form can also affect adolescents and adults. Persons with the ALD mutation may also be detected in an asymptomatic or presymptomatic stage during screening of at-risk family members.
The cerebral form is the most common subtype in children and begins between ages 4 and 8, with a mean age of onset of 7.2 years.4 Affected children first develop symptoms suggestive of attention deficit hyperactivity disorder, which are followed by signs of impaired auditory discrimination, cognitive dysfunction, visual abnormalities, ataxia, and, later, signs of corticospinal tract involvement. Seizures occasionally may be the heralding event. Adrenal function is abnormal in 90% of patients.4 After the initial onset, the illness advances rapidly to a vegetative state within a mean of 1.9 years, with death occurring at a mean age of 9.4 years.8
Adrenomyeloneuropathy (AMN) is the most common adult form, with a mean age of onset of 27 years. The main clinical findings are a slowly progressive spastic paraparesis, with sensory disturbances most severe in the distal aspects of the lower extremities and sphincter disturbances. These symptoms reflect the predominant involvement of the spinal cord and peripheral nerves in these patients. Impotence develops in the later stages, although many of these male patients have fathered children. Brain involvement occurs in 40% to 45% of patients with AMN based on clinical exam or MRI and is clinically severe in 10% to 20%.4 The AMN subtype also affects 20% of heterozygote females who develop milder symptoms in the third to fifth decades.
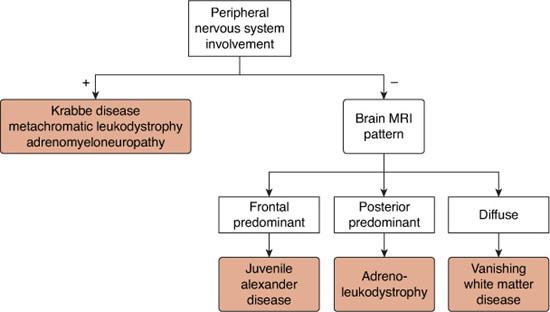
FIGURE 576-2. Algorithm for the differential diagnosis of leukodystrophies in childhood with onset after the first year of life.
 DIAGNOSIS
DIAGNOSIS
The initial differential diagnosis of the childhood cerebral form of adrenoleukodystrophy (ALD) mainly includes distinguishing it from primary attention deficit hyperactivity disorder. As symptoms progress, ALD should be distinguished from other childhood-onset leukodystrophies, such as juvenile-onset metachromatic leukodystrophy, on the basis of clinical and MRI findings, with laboratory confirmation. Patients with ALD who present acutely with gadolinium enhancement may be thought to have acquired demyelinating disorders, such as acute disseminated encephalomyelitis or multiple sclerosis; these disorders tend to have much more asymmetric MRI abnormalities. Disorders that can mimic the AMN subtype include multiple sclerosis, vitamin B12 deficiency, and hereditary spastic paraparesis.
The diagnosis of ALD can be confirmed by the detection of elevated levels of very long chain fatty acids (VLCFA) in plasma, particularly C26:0 (hexacosanoic acid), the ratio of C24:0 to C22:0, and the ratio of C26:0 to C22:0. These values are elevated in 99.9% of affected males and 85% of carrier females.4 Although sequencing of the ABCD1 gene can be performed, it is clinically useful only in rare male patients with nondiagnostic VLCFA values and carrier females with normal VLCFA levels.
Gadolinium-enhanced brain MRI should be performed in all patients. Brain MRI shows preferential, symmetric involvement of the parieto-occipital white matter and/or splenium of the corpus callosum in 66% of patients overall and 80% of patients under 10 years of age (Fig. 576-3).9 When present, gadolinium enhancement is typically curvilinear and close to the advancing margin, correlating with the middle zone on neuropathology specimens. Enhancement is found in approximately 45% of patients overall and predicts faster clinical and radiographic progression.9
 MANAGEMENT
MANAGEMENT
All patients should undergo an assessment of their adrenal function with measurement of plasma adrenocorticotropin hormone (ACTH) levels or cortisol response in an ACTH stimulation test with the assistance of an endocrinologist. Patients with initially normal results should be reevaluated regularly. The first priority of treatment in ALD is to provide adrenal hormone supplementation, if adrenal insufficiency is identified, in order to avoid the morbidity and mortality associated with adrenal crises.
Additional treatment of ALD may include Lorenzo’s oil, a 4:1 mixture of glycerol trioleate and glycerol trierucate that lowers plasma VLCFA levels by approximately 50% in patients with ALD, likely via competitive inhibition of VLCFA synthesis.6 Although it is ineffective in significantly altering clinical progression in patients with cerebral ALD with established neurologic symptoms, it may have a role in disease prevention. In an open-label, single-arm trial involving 89 boys with ALD, no neurologic symptoms, and a normal brain MRI, Lorenzo’s oil therapy appeared to reduce the risk of developing the childhood cerebral form of the disease by twofold over a mean follow-up of 6.9 years. In 30% to 40% of treated patients, Lorenzo’s oil reduces platelet counts, which should be followed regularly. Very close nutritional and biochemical monitoring must also be carried out.
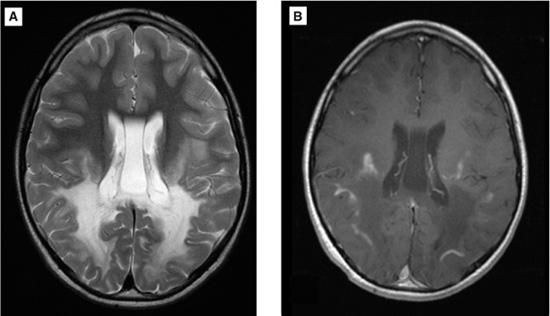
FIGURE 576-3. Brain MRI of an 8-year-old boy who developed declining school performance, clumsiness, and visual impairment at age 5 years. His symptoms became progressively worse over time. Physical exam showed cognitive impairment and hyperreflexia. Plasma levels of very-long-chain fatty acids were increased, confirming a diagnosis of X-linked adrenoleukodystrophy. A: Axial T2-weighted image showing symmetric, posterior-predominant hyperintense signal. B: Axial, postgadolinium T1-weighted image showing T1 hypointensity in the region of T2 hyperintensity. Contrast enhancement is present at the peripheral margin. (Courtesy of Dr. Omar Khwaja, Department of Neurology, Children’s Hospital, Boston, MA.)
Hematopoietic stem cell transplantation (HSCT) improves survival and neurologic function in patients with childhood cerebral ALD, particularly those early in the disease course. Although overall 5-year survival was 56% in 94 patients with ALD who received HSCT and 66% in a historical cohort of non-transplanted patients, this figure was significantly higher in patients with mild neurologic deficits and/or MRI findings at the time of HSCT.10,11 In a subgroup of 19 patients with 0 or 1 neurologic deficits and MRI severity score less than 9 on a standardized scale, the 5-year survival rate was 95% compared to 54% in a historical cohort of 30 nontransplanted patients with a similarly mild degree of neurologic deficits.10,11 In addition, 53% of the surviving transplanted patients in this subgroup were neurologically stable at 5 years, compared to only 6% in the historical control group.11 Patients with more significant clinical and MRI disease burden are at high risk of death following HSCT, which is therefore not recommended for this group of patients. In addition, as only 35% to 40% of boys younger than 10 years old with biochemical evidence of ALD will develop childhood cerebral ALD and there are substantial risks associated with HSCT, this treatment is not recommended for patients without MRI evidence of demyelination.2 Therefore, the challenge is to identify the patients who have minimal, but not more advanced, neurologic involvement and to successfully perform HSCT in them quickly. The mechanism by which HSCT affects ALD is uncertain, but probably involves alterations in the immune system, rather than replacement of the defective gene product.
 OUTCOME AND PREVENTION
OUTCOME AND PREVENTION
As described above, the prognosis for ALD depends on the specific clinical phenotype. Within the childhood cerebral subtype, the overall prognosis is poor in the absence of timely HSCT. Unfortunately, plasma levels of VLCFA and specific mutations in ABCD1 do not correlate well with the clinical phenotype, making prognostication difficult. Patients with the same mutation within the same family can have different clinical expression.  Early age of onset is also associated with a worse prognosis.9
Early age of onset is also associated with a worse prognosis.9
The diagnosis of ALD can now be made via analysis of newborn blood spots, thus making newborn screening a possibility. It is estimated that at least 50 presymptomatic boys with ALD would be identified each year in the United States.6 Importantly, 92% of boys with ALD with normal brain MRI have normal cognition on detailed neuropsychological testing.12 This finding, combined with the potential efficacy of Lorenzo’s oil in delaying disease expression and the proven efficacy of HSCT in early-stage patients, makes newborn screening an attractive, although challenging, possibility. Early diagnosis would substantially increase the currently narrow therapeutic window of opportunity by allowing advanced preparation for HSCT should it be needed. In families in which the specific mutation is known, prenatal diagnosis is also available.
AICARDI-GOUTIERES SYNDROME
Patients with Aicardi-Goutieres syndrome develop a subacute, severe encephalopathy within the first year of life, with irritability, developmental regression, and slowing of head growth. This phase usually lasts several months, after which the patient has severe cognitive impairment, spasticity, and microcephaly, but does not continue to regress.13 More than 50% also have seizures and 40% have chilblain lesions on the digits or ears. Fever in the absence of infection is also common. Computerized tomography of the head shows calcification of the basal ganglia and periventricular white matter. Brain MRI reveals hyperintense signal in the periventricular white matter with a frontal predominance on T2-weighted images. Cerebrospinal fluid contains elevated levels of white blood cells, interferon-α, and neopterin, which gradually decline over several years. Mutations in AGS1, 2, 3, or 4 cause approximately 80% of cases of the syndrome, with nearly all patients having homozygous recessive or compound heterozygote mutations.13AGS1 encodes for 3 prime repair exonuclease 1 (TREX1). Mutations in this gene can produce the syndrome at birth, with associated hepatosplenomegaly, transaminitis, and thrombocytopenia. AGS2, 3, and 4 encode for subunits of ribonuclease H. Due to the presence of neurologic abnormalities, microcephaly, and intracranial calcifications, congenital toxoplasmosis, rubella, CMV, and herpes simplex (TORCH) infections should be ruled out with appropriate serologic and virologic studies in patients suspected of having Aicardi-Goutieres syndrome.
ALEXANDER DISEASE
Alexander disease is a rare, autosomal dominant leukodystrophy that has been reported in many different racial and ethnic groups. Its exact incidence is uknown.14
Prior to 2001, the definitive diagnosis of Alexander disease required the demonstration of Rosenthal fibers, eosinophilic rodlike intracellular inclusions composed of aggregates of glial fibrillary acidic protein (GFAP) and other proteins, on brain biopsy specimens of affected patients. In 2001, Brenner and colleagues discovered mutations in GFAP on chromosome 17q21 in 10 of 11 patients with Alexander disease.15 Subsequent studies have shown that GFAP mutations account for more than 90% of all cases of Alexander disease.16 Nearly all of the identified mutations are missense mutations involving exons 1, 4, or 6, with hotspots at residues R79 and R239.17 GFAP is the major intermediate filament protein in CNS astrocytes. The exact mechanisms by which alterations in GFAP in astrocytes lead to the myelin abnormalities seen in Alexander disease are unknown.
Three forms of Alexander disease are recognized based on age of onset. The infantile-onset form manifests within the first 2 years of life and comprises approximately 65% of cases.14 Typical symptoms include developmental delay, megalencephaly, and seizures. As the disease progresses, cognitive decline, feeding problems, and spastic quadriparesis become apparent. Some infants present acutely in the first year with hydrocephalus and increased intracranial pressure due to aqueductal stenosis. Death usually occurs between the ages of 2 and 10 years. A more fulminant neonatal form with onset in the first month of life and very rapid progression has also been proposed.
Juvenile-onset Alexander disease (25%) usually begins between the ages of 4 and 10 years. The most common symptoms consist of bulbar or pseudobulbar dysfunction, especially dysphagia. Spasticity, ataxia, and cognitive regression may also occur. Megalencephaly may occur, but is less common than in the infantile-onset form. These children may survive for up to 10 years and frequently longer. Adult-onset forms comprise 10% of all cases with marked variability in presentation.14
The combination of macrocephaly and leukodystrophy in the first 2 years of life is suggestive of Alexander disease. MRI findings and biochemical and genetic testing help to differentiate it from other disorders such as Canavan disease and megalencephalic leukoencephalopathy with subcortical cysts. Juvenile-onset cases can be distinguished from other leukodystrophies with a similar age of onset, such as the juvenile-onset forms of metachromatic leukodystrophy and Krabbe disease, by the lack of peripheral nervous system involvement, as well as MRI features.
MRI in Alexander disease demonstrates extensive, symmetric T2 hyperintense signal in the cerebral white matter with predominant involvement of the frontal lobes (Fig. 576-4). Additional common findings include (1) a periventricular rim of T1 hyper-intensity and T2 hypointensity, (2) swelling and later atrophy of the basal ganglia and thalamus, (3) brainstem abnormalities, and (4) contrast enhancement involving various locations. The presence of 4 of 5 of these common findings makes the diagnosis of Alexander disease likely, based on a large, multicenter study.18 However, although frontal-predominant white matter T2 hyperintensities remains the most classic finding, recent studies have demonstrated significant heterogeneity in the MRI appearance.19 Thus, patients suspected of having Alexander disease should undergo sequencing of the GFAP gene, even in the presence of atypical MRI findings.
There is no specific treatment for Alexander disease, although supportive treatment may improve symptoms. The overall prognosis is poor, although significant differences in rates of progression and age at death exist between the different subtypes. Due to its relative rarity, predictive genotype-phenotype correlations do not exist. Prenatal diagnosis is possible in families in which the disease-causing mutation is known.
CANAVAN DISEASE
Canavan disease is an autosomal-recessive neurologic disorder that affects all ethnic groups, but has its highest rate in Ashkenazi Jews. In this group, the carrier rates have been estimated between 1:40 and 1:60.20
Mutations in the ASPA gene on chromosome 17p cause Canavan disease. This gene encodes aspartoacylase, an enzyme that is responsible for hydrolyzing N-acetylaspartic acid to aspartic acid and acetate. Two mutations (E285A and Y231X) account for 98% of alleles in Ashkenazi Jewish patients, while 1 mutation (A305E) is found in approximately 50% of non-Jewish patients.20 Deficiency of the enzyme leads to accumulation of N-acetylaspartic acid in the brain, which in turn causes spongiform degeneration of the white matter via unknown mechanisms.
Following normal initial development, patients with Canavan disease develop the classic triad of macrocephaly, hypotonia, and poor head control.23 Delayed development is apparent by 3 months of age. Macrocephaly may not be apparent within the first few months, but the head enlarges to above the 90th percentile within 6 months to a year of life. Affected children never develop the ability to support the head, sit, or stand independently. Seizures and optic atrophy frequently develop in the second year of life. Over time, hypotonia evolves into spasticity, gastroesophageal reflux becomes prominent, and swallowing deteriorates, leading to feeding difficulties, poor weight gain, and need for gastrostomy tube feeding.20 The rate of overall disease progression is variable.23
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


